
27.885 personas tomaron semaglutida o tirzepatida. Una sola variante en tu ADN puede triplicar el riesgo de vómitos — pero solo con uno de los dos fármacos.
Paper: Genetic predictors of GLP1 receptor agonist weight loss and side effects DOI: 10.1038/s41586-026-10330-z Datos: Supplementary Data (Nature)
![]()
El problema de la variabilidad#
Ozempic, Wegovy, Mounjaro, Zepbound — los agonistas del receptor GLP1 transformaron el tratamiento de la obesidad. Pero la respuesta varía enormemente: hay quien pierde el 20% de su peso corporal y quien apenas baja un 2%.
Un equipo de 23andMe reunió a 27.885 personas en tratamiento con estos fármacos y buscó en su genoma qué variantes predicen dos cosas: cuánto peso pierdes y qué efectos secundarios sufres. Lo que encontraron sienta las bases para la medicina de precisión en obesidad.
# ==============================================================
# Configuración — modifica estos valores para explorar
# ==============================================================
FUENTE = 'Fuente: 23andMe et al. (2026), Nature | Supplementary Data'
COLOR_TIRZ = '#2563EB'
COLOR_SEMA = '#059669'
COLOR_ALERTA = '#DC2626'
COLOR_REF = '#D97706'
COLOR_VIOLETA = '#7C3AED'
COLOR_GRIS = '#BBBBBB'
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import os, urllib.request
style_file = '../../cam.mplstyle'
if not os.path.exists(style_file):
style_file = '/tmp/cam.mplstyle'
if not os.path.exists(style_file):
urllib.request.urlretrieve(
'https://raw.githubusercontent.com/Ciencia-a-Mordiscos/lab/main/cam.mplstyle',
style_file)
plt.style.use(style_file)
efectos = pd.read_csv('datos/efectos_genotipo.csv')
farmacos = pd.read_csv('datos/resumen_farmacos.csv')
variantes = pd.read_csv('datos/variantes_gwas.csv')
modelo = pd.read_csv('datos/modelo_eficacia.csv')
rendimiento = pd.read_csv('datos/rendimiento_modelo.csv')
gwas_peso = pd.read_csv('datos/gwas_peso_cambio.csv')
gwas_fenotipos = pd.read_csv('datos/gwas_resumen_fenotipos.csv')
print(f"Participantes: 27.885 (4.908 hombres + 22.977 mujeres)")
print(f"Variantes GWAS significativas: {len(variantes)}")
print(f"SNPs con p < 1x10-4 (cambio de peso): {len(gwas_peso)}")
print(f"Fenotipos analizados: {len(gwas_fenotipos)}")
print()
print("Fármacos: Ozempic, Wegovy, Mounjaro, Zepbound + compuestos")
Participantes: 27.885 (4.908 hombres + 22.977 mujeres)
Variantes GWAS significativas: 9
SNPs con p < 1x10-4 (cambio de peso): 1626
Fenotipos analizados: 39
Fármacos: Ozempic, Wegovy, Mounjaro, Zepbound + compuestos
Una variante, dos fármacos, tres veces más riesgo#
Aquí está.
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(13, 5.5), sharey=True)
genotipos = ['CC', 'CG', 'GG']
x = np.arange(len(genotipos))
width = 0.35
# Panel izquierdo: Tirzepatida
vom_tirz = efectos[(efectos['efecto'] == 'vomiting') & (efectos['farmaco'] == 'tirzepatide')]
nau_tirz = efectos[(efectos['efecto'] == 'nausea') & (efectos['farmaco'] == 'tirzepatide')]
vom_vals_t = [vom_tirz[vom_tirz['genotipo'] == g]['prevalencia_pct'].values[0] for g in genotipos]
nau_vals_t = [nau_tirz[nau_tirz['genotipo'] == g]['prevalencia_pct'].values[0] for g in genotipos]
bars1 = ax1.bar(x - width/2, vom_vals_t, width, color=COLOR_ALERTA, alpha=0.85,
label='Vómitos', zorder=5)
bars2 = ax1.bar(x + width/2, nau_vals_t, width, color=COLOR_TIRZ, alpha=0.85,
label='Náusea', zorder=5)
for bar, val in zip(bars1, vom_vals_t):
ax1.text(bar.get_x() + bar.get_width()/2, bar.get_height() + 0.5,
f'{val}%', ha='center', va='bottom', fontsize=9, fontweight='bold',
color=COLOR_ALERTA)
for bar, val in zip(bars2, nau_vals_t):
ax1.text(bar.get_x() + bar.get_width()/2, bar.get_height() + 0.5,
f'{val}%', ha='center', va='bottom', fontsize=9, fontweight='bold',
color=COLOR_TIRZ)
ax1.set_title('Tirzepatida', fontsize=14, fontweight='bold', pad=20, color=COLOR_TIRZ)
ax1.text(0.5, 1.02, 'El genotipo cambia el riesgo',
transform=ax1.transAxes, fontsize=10, color='#666666', ha='center')
ax1.set_xticks(x)
ax1.set_xticklabels(genotipos, fontsize=11, fontweight='bold')
ax1.set_ylabel('Prevalencia (%)', fontsize=11)
ax1.legend(fontsize=9, loc='upper right', framealpha=0.9)
ax1.set_ylim(0, 33)
ax1.annotate('', xy=(0 - width/2, vom_vals_t[0] + 2.5),
xytext=(2 - width/2, vom_vals_t[2] + 2.5),
arrowprops=dict(arrowstyle='<->', color='#666666', lw=1.5))
ax1.text(1 - width/2, max(vom_vals_t[0], vom_vals_t[2]) + 3.5, '3,0x',
ha='center', fontsize=11, fontweight='bold', color=COLOR_ALERTA)
# Panel derecho: Semaglutida
vom_sema = efectos[(efectos['efecto'] == 'vomiting') & (efectos['farmaco'] == 'semaglutide')]
nau_sema = efectos[(efectos['efecto'] == 'nausea') & (efectos['farmaco'] == 'semaglutide')]
vom_vals_s = [vom_sema[vom_sema['genotipo'] == g]['prevalencia_pct'].values[0] for g in genotipos]
nau_vals_s = [nau_sema[nau_sema['genotipo'] == g]['prevalencia_pct'].values[0] for g in genotipos]
bars3 = ax2.bar(x - width/2, vom_vals_s, width, color=COLOR_ALERTA, alpha=0.85, zorder=5)
bars4 = ax2.bar(x + width/2, nau_vals_s, width, color=COLOR_SEMA, alpha=0.85, zorder=5)
for bar, val in zip(bars3, vom_vals_s):
ax2.text(bar.get_x() + bar.get_width()/2, bar.get_height() + 0.5,
f'{val}%', ha='center', va='bottom', fontsize=9, fontweight='bold',
color=COLOR_ALERTA)
for bar, val in zip(bars4, nau_vals_s):
ax2.text(bar.get_x() + bar.get_width()/2, bar.get_height() + 0.5,
f'{val}%', ha='center', va='bottom', fontsize=9, fontweight='bold',
color=COLOR_SEMA)
ax2.set_title('Semaglutida', fontsize=14, fontweight='bold', pad=20, color=COLOR_SEMA)
ax2.text(0.5, 1.02, 'El genotipo NO cambia el riesgo',
transform=ax2.transAxes, fontsize=10, color='#666666', ha='center')
ax2.set_xticks(x)
ax2.set_xticklabels(genotipos, fontsize=11, fontweight='bold')
ax2.text(1, max(vom_vals_s) + 5, 'p = 0,70',
ha='center', fontsize=10, color='#999999', style='italic')
fig.text(0.5, -0.02, 'Genotipo GIPR rs1800437', ha='center', fontsize=11, fontweight='bold')
fig.text(0.13, -0.06, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.tight_layout()
plt.savefig('figuras/interaccion_gen_farmaco.png', dpi=200, bbox_inches='tight')
plt.show()
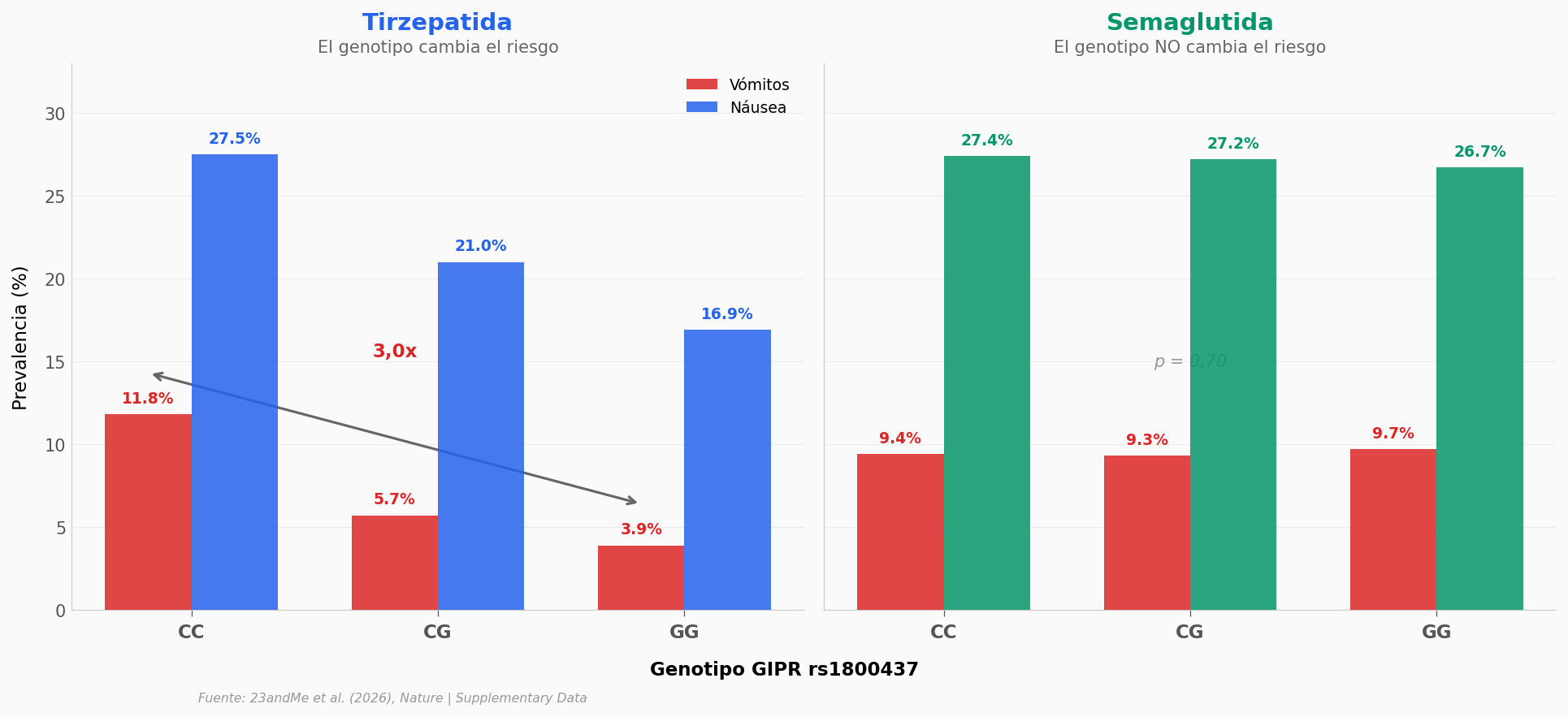
El gradiente del panel izquierdo es inequívoco. En personas que toman tirzepatida (Mounjaro, Zepbound), el genotipo CC del gen GIPR se asocia con un 11,8% de prevalencia de vómitos — el triple que el 3,9% del genotipo GG. El test de tendencia entre los tres genotipos (CC, CG, GG) confirma la asociación (chi-cuadrado = 44,5, p = 2,2 x 10 a la -10). La náusea sigue el mismo patrón: 27,5% vs 16,9% (p = 4,6 x 10 a la -8).
El panel derecho lo pone en perspectiva. Para semaglutida, la misma variante genética no tiene ningún efecto (p = 0,70 para vómitos). Los tres genotipos rondan el 9,4%.
¿Por qué? Tirzepatida actúa sobre DOS receptores: GLP1R y GIPR. La variante rs1800437 está en el gen GIPR. Si tu GIPR tiene el genotipo CC, tirzepatida activa una señal gastrointestinal que semaglutida — que solo toca GLP1R — no activa.
¿Y la pérdida de peso?#
Los efectos secundarios son la mitad de la historia. La otra es cuánto peso se pierde — y qué fármaco lo logra.
fig, ax = plt.subplots(figsize=(13, 5.5))
drugs_order = ['Mounjaro', 'Zepbound', 'Compounded_tirzepatide',
'Wegovy', 'Ozempic', 'Compounded_semaglutide']
drug_labels = ['Mounjaro (tirzepatida)', 'Zepbound (tirzepatida)',
'Compuesta (tirzepatida)',
'Wegovy (semaglutida)', 'Ozempic (semaglutida)',
'Compuesta (semaglutida)']
y_pos = np.arange(len(drugs_order))
bar_height = 0.35
for i, drug in enumerate(drugs_order):
for sex, offset, alpha, marker in [('F', -bar_height/2, 0.85, chr(9792)),
('M', bar_height/2, 0.6, chr(9794))]:
row = farmacos[(farmacos['farmaco'] == drug) & (farmacos['sexo'] == sex)]
if len(row) == 0:
continue
bmi_str = row['bmi_change_pct_mediana'].values[0]
val = float(bmi_str.split('%')[0])
n = int(row['n'].values[0])
is_tirz = drug in ['Mounjaro', 'Zepbound', 'Compounded_tirzepatide']
color = COLOR_TIRZ if is_tirz else COLOR_SEMA
ax.barh(i + offset, abs(val), height=bar_height,
color=color, alpha=alpha, zorder=5)
n_str = f'{n:,}'.replace(',', '.')
ax.text(abs(val) + 0.2, i + offset,
f'{val}% {marker} (n={n_str})',
va='center', fontsize=8.5, color=color, fontweight='bold')
ax.set_yticks(y_pos)
ax.set_yticklabels(drug_labels, fontsize=9)
ax.set_xlabel('Reducción mediana del IMC (%)', fontsize=11)
ax.set_title('¿Qué fármaco reduce más el IMC?', fontsize=14, fontweight='bold', pad=20)
ax.text(0.5, 1.02,
'Mediana del % de cambio de IMC por fármaco y sexo (27.885 participantes)',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.axhline(y=2.5, color='#DDDDDD', linewidth=1, linestyle='--')
ax.text(17, 1, 'Tirzepatida', fontsize=10, fontweight='bold',
color=COLOR_TIRZ, ha='center', va='center')
ax.text(17, 4, 'Semaglutida', fontsize=10, fontweight='bold',
color=COLOR_SEMA, ha='center', va='center')
ax.invert_yaxis()
ax.set_xlim(0, 19)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/comparacion_farmacos.png', dpi=200, bbox_inches='tight')
plt.show()
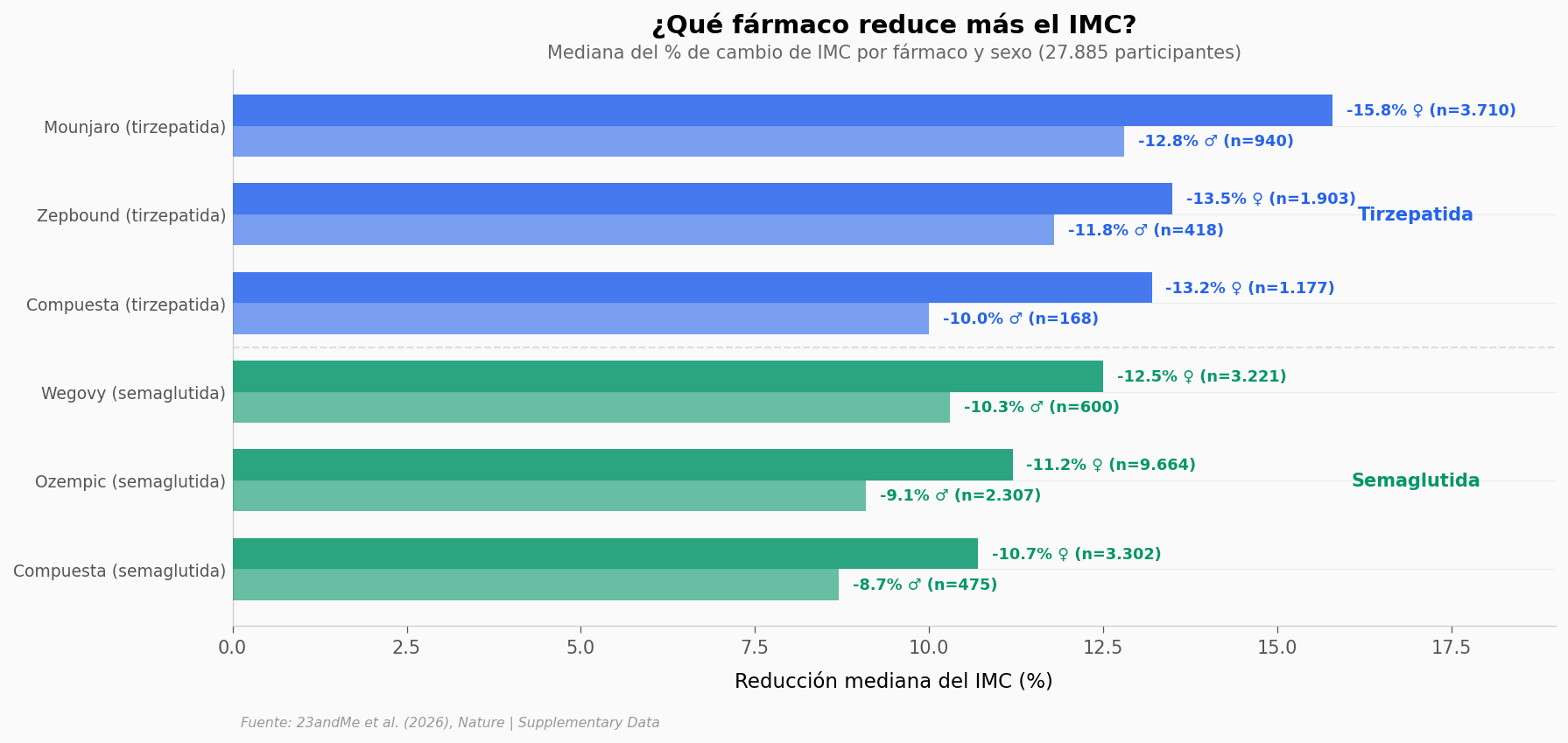
Mounjaro lidera: las mujeres que lo toman alcanzan una mediana de -15,8% de IMC. Los hombres, -12,8%. Tirzepatida supera a semaglutida en casi todas las comparaciones (excepción: tirzepatida compuesta vs Wegovy en hombres, -10,0% vs -10,3%).
Pero esas medianas ocultan una dispersión enorme. ¿Qué lo explica?
Un modelo de medicina de precisión#
El equipo construyó un modelo que combina factores clínicos y genéticos para predecir la respuesta. Veamos qué pesa más.
fig, ax = plt.subplots(figsize=(13, 6))
main_vars = ['const', 'Dose', 'Drug Type', 'T2D', 'Female Sex', 'NAFLD',
'GWAS Index SNP*', 'Hypertension', 'Years of Education', 'Age']
labels_es = ['Constante', 'Dosis', 'Tipo de fármaco (tirz vs sema)',
'Diabetes tipo 2', 'Sexo femenino', 'Hígado graso',
'Variante genética (rs10305420)', 'Hipertensión',
'Años de educación', 'Edad']
model_main = modelo[modelo['variable'].isin(main_vars)].copy()
model_main['order'] = model_main['variable'].map(
{v: i for i, v in enumerate(main_vars)})
model_main = model_main.sort_values('order')
coefs = model_main['coef'].values
stderrs = model_main['stderr'].values
y = np.arange(len(coefs))
colors = [COLOR_TIRZ if c < 0 else COLOR_ALERTA for c in coefs]
bars = ax.barh(y, coefs, xerr=stderrs * 1.96, height=0.6,
color=colors, alpha=0.75, capsize=3, zorder=5,
error_kw={'linewidth': 1, 'color': '#666666'})
ax.axvline(x=0, color='#333333', linewidth=0.8, zorder=3)
snp_idx = list(main_vars).index('GWAS Index SNP*')
bars[snp_idx].set_edgecolor(COLOR_VIOLETA)
bars[snp_idx].set_linewidth(2)
ax.set_yticks(y)
ax.set_yticklabels(labels_es, fontsize=9)
ax.set_xlabel('Coeficiente (% cambio IMC)', fontsize=11)
ax.set_title('¿Qué predice cuánto peso pierdes?',
fontsize=14, fontweight='bold', pad=20)
ax.text(0.5, 1.02,
'Modelo combinado: factores clínicos + genéticos (n = 11.151)',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.text(-6.134, 1.15, chr(8592) + ' mas perdida', fontsize=8,
color=COLOR_TIRZ, fontweight='bold')
ax.text(2.752, 3.15, 'menos perdida ' + chr(8594), fontsize=8,
color=COLOR_ALERTA, fontweight='bold')
ax.text(0.98, 0.02, 'barras = IC 95%', transform=ax.transAxes,
fontsize=8, color='#999999', ha='right', va='bottom', style='italic')
ax.invert_yaxis()
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/modelo_eficacia.png', dpi=200, bbox_inches='tight')
plt.show()
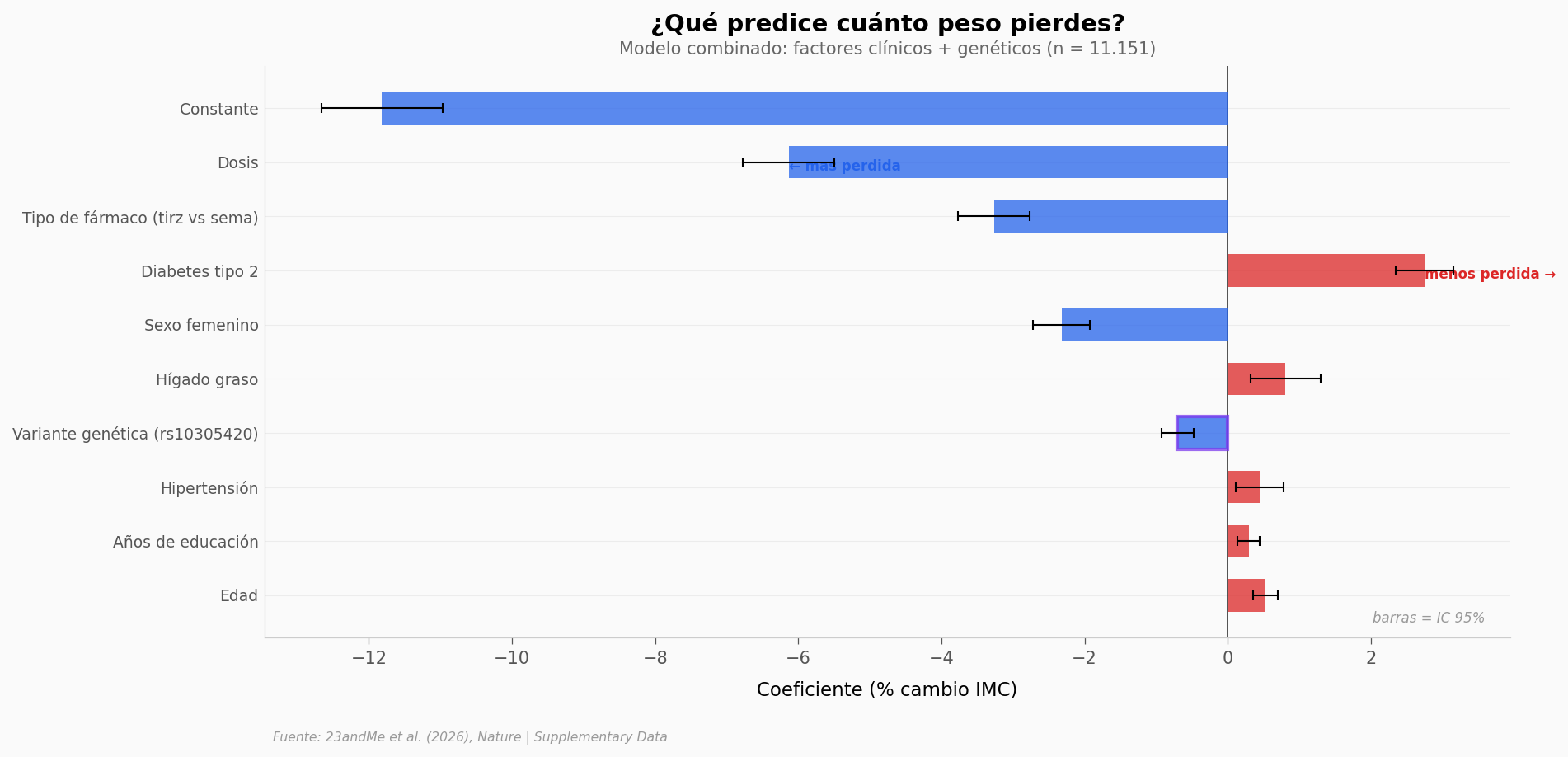
La dosis y el tipo de fármaco dominan — eran esperables. Pero fijémonos en tres hallazgos:
Sexo femenino (coef = -2,32): las mujeres pierden más peso en promedio, un efecto más grande que la variante genética.
Diabetes tipo 2 (coef = +2,75): los diabéticos pierden menos, probablemente porque la indicación original (control glucémico) compite con el efecto de pérdida de peso.
Variante genética rs10305420 (coef = -0,70): cada copia del alelo T se asocia con un 0,70% adicional de reducción de IMC (p = 7,9 x 10 a la -10). Es más modesto que el tipo de fármaco, pero es la primera evidencia directa de que una variante en el gen diana contribuye a la variabilidad.
¿Puede la genética mejorar las predicciones?#
El modelo con genética versus sin genética — ¿cuánto mejora?
fig, ax = plt.subplots(figsize=(13, 5.5))
side_effects = rendimiento[rendimiento['type'] == 'side_effect'].copy()
side_effects = side_effects.sort_values('auc_without_genetics')
phenotype_labels = {
'muscle weakness': 'Debilidad muscular',
'constipation': 'Estreñimiento',
'pleasure eating': 'Placer al comer',
'taste': 'Cambios de sabor',
'stomach pain': 'Dolor estomacal',
'appetite': 'Apetito',
'diarrhea': 'Diarrea',
'nausea': 'Náusea',
'vomiting': 'Vómitos',
'butt sagging': 'Flacidez glútea',
'face sagging': 'Flacidez facial',
}
labels = [phenotype_labels.get(p, p) for p in side_effects['phenotype']]
y = np.arange(len(labels))
auc_no_gen = side_effects['auc_without_genetics'].values
bars = ax.barh(y, auc_no_gen, height=0.6, color=COLOR_GRIS, alpha=0.6,
zorder=4, label='Sin genética')
for i, (_, row) in enumerate(side_effects.iterrows()):
if pd.notna(row['auc_with_genetics']):
delta = row['auc_with_genetics'] - row['auc_without_genetics']
ax.barh(i, row['auc_with_genetics'], height=0.6,
color=COLOR_VIOLETA, alpha=0.75, zorder=5)
ax.text(row['auc_with_genetics'] + 0.005, i,
f"+{delta:.3f}",
va='center', fontsize=9, fontweight='bold', color=COLOR_VIOLETA)
ax.axvline(x=0.5, color=COLOR_ALERTA, linewidth=1.5, linestyle='--',
alpha=0.7, zorder=3)
ax.text(0.502, len(labels) - 0.5, 'Azar', fontsize=9,
color=COLOR_ALERTA, fontweight='bold')
ax.set_yticks(y)
ax.set_yticklabels(labels, fontsize=9)
ax.set_xlabel('AUC (Área Bajo la Curva ROC)', fontsize=11)
ax.set_title('¿Qué efectos secundarios podemos predecir?',
fontsize=14, fontweight='bold', pad=20)
ax.text(0.5, 1.02,
'Rendimiento del modelo — barras violeta = mejora al agregar genética',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.set_xlim(0.49, 0.73)
from matplotlib.patches import Patch
legend_elements = [
Patch(facecolor=COLOR_GRIS, alpha=0.6, label='Solo factores clínicos'),
Patch(facecolor=COLOR_VIOLETA, alpha=0.75, label='Clínicos + genética')]
ax.legend(handles=legend_elements, fontsize=9, loc='lower right', framealpha=0.9)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/rendimiento_modelos.png', dpi=200, bbox_inches='tight')
plt.show()
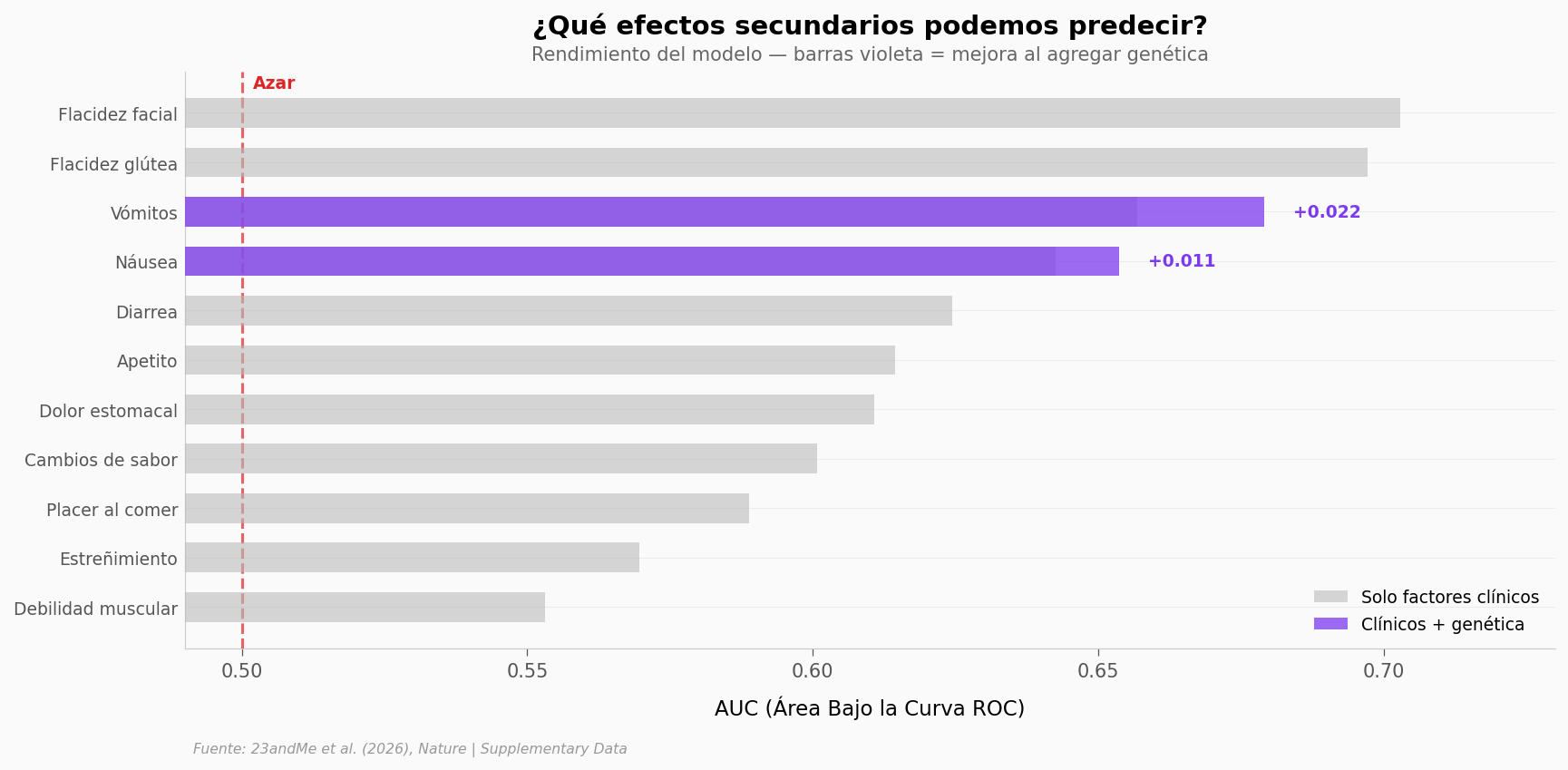
Vómitos y náusea son los únicos efectos secundarios donde la genética mejora la predicción (delta AUC = +0,022 y +0,011). Para los demás, la genética disponible no aporta.
La flacidez facial y glútea tienen los AUC más altos (~0,70), pero eso viene de factores clínicos (tiempo en tratamiento, cantidad de peso perdido), no de genética.
Para la eficacia (% de pérdida de IMC), el R cuadrado del modelo es 0,243 con genética y 0,242 sin ella — una mejora de 0,001. La variante rs10305420 aporta, pero la mayor parte de la variabilidad la explican dosis, tipo de fármaco y condiciones preexistentes.
¿Dónde están las variantes?#
fig, ax = plt.subplots(figsize=(13, 5))
gwas = gwas_peso.copy()
# Map chromosome names to numbers (X=23, Y=24, MT=25)
chr_map = {'X': 23, 'Y': 24, 'MT': 25}
gwas['chr_num'] = gwas['CHR'].apply(lambda c: chr_map.get(str(c), int(c) if str(c).isdigit() else None))
gwas = gwas.dropna(subset=['chr_num'])
gwas['chr_num'] = gwas['chr_num'].astype(int)
gwas['neg_log_p'] = -np.log10(gwas['p-value'])
chr_colors = {c: (COLOR_TIRZ if c % 2 == 0 else COLOR_SEMA)
for c in gwas['chr_num'].unique()}
colors = [chr_colors[c] for c in gwas['chr_num']]
chr_offset = {}
cumulative = 0
for chrom in sorted(gwas['chr_num'].unique()):
chr_offset[chrom] = cumulative
sub = gwas[gwas['chr_num'] == chrom]
cumulative += sub['POS_b38'].max() - sub['POS_b38'].min() + 5e7
gwas['plot_pos'] = gwas.apply(
lambda r: chr_offset[r['chr_num']] + r['POS_b38'], axis=1)
ax.scatter(gwas['plot_pos'], gwas['neg_log_p'], c=colors,
s=12, alpha=0.6, edgecolors='none', zorder=4)
gw_threshold = -np.log10(5e-8)
ax.axhline(y=gw_threshold, color=COLOR_ALERTA, linewidth=1.2,
linestyle='--', alpha=0.7, zorder=3)
ax.text(gwas['plot_pos'].max() * 0.98, gw_threshold + 0.3,
'p = 5x10\u207b\u2078', fontsize=8,
color=COLOR_ALERTA, ha='right', fontweight='bold')
top_snp = gwas.loc[gwas['neg_log_p'].idxmax()]
ax.annotate("rs10305420 (GLP1R)", xy=(top_snp['plot_pos'], top_snp['neg_log_p']),
xytext=(top_snp['plot_pos'] + cumulative * 0.08,
top_snp['neg_log_p'] - 0.5),
fontsize=9, fontweight='bold', color=COLOR_ALERTA,
arrowprops=dict(arrowstyle='->', color=COLOR_ALERTA, lw=1.2))
chr_labels_map = {23: 'X', 24: 'Y', 25: 'MT'}
chr_centers = {}
for chrom in sorted(gwas['chr_num'].unique()):
positions = gwas[gwas['chr_num'] == chrom]['plot_pos']
chr_centers[chrom] = (positions.min() + positions.max()) / 2
ax.set_xticks(list(chr_centers.values()))
ax.set_xticklabels([chr_labels_map.get(c, str(c)) for c in chr_centers.keys()], fontsize=7)
ax.set_xlabel('Cromosoma', fontsize=11)
ax.set_ylabel('\u2212log\u2081\u2080(p)', fontsize=11)
ax.set_title('¿Dónde están las variantes que importan?',
fontsize=14, fontweight='bold', pad=20)
n_gw = len(gwas[gwas['neg_log_p'] >= gw_threshold])
subtitle = f'{len(gwas)} SNPs con p < 1x10\u207b\u2074 \u2014 {n_gw} alcanzan significancia genómica'
ax.text(0.5, 1.02, subtitle,
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/manhattan_peso.png', dpi=200, bbox_inches='tight')
plt.show()
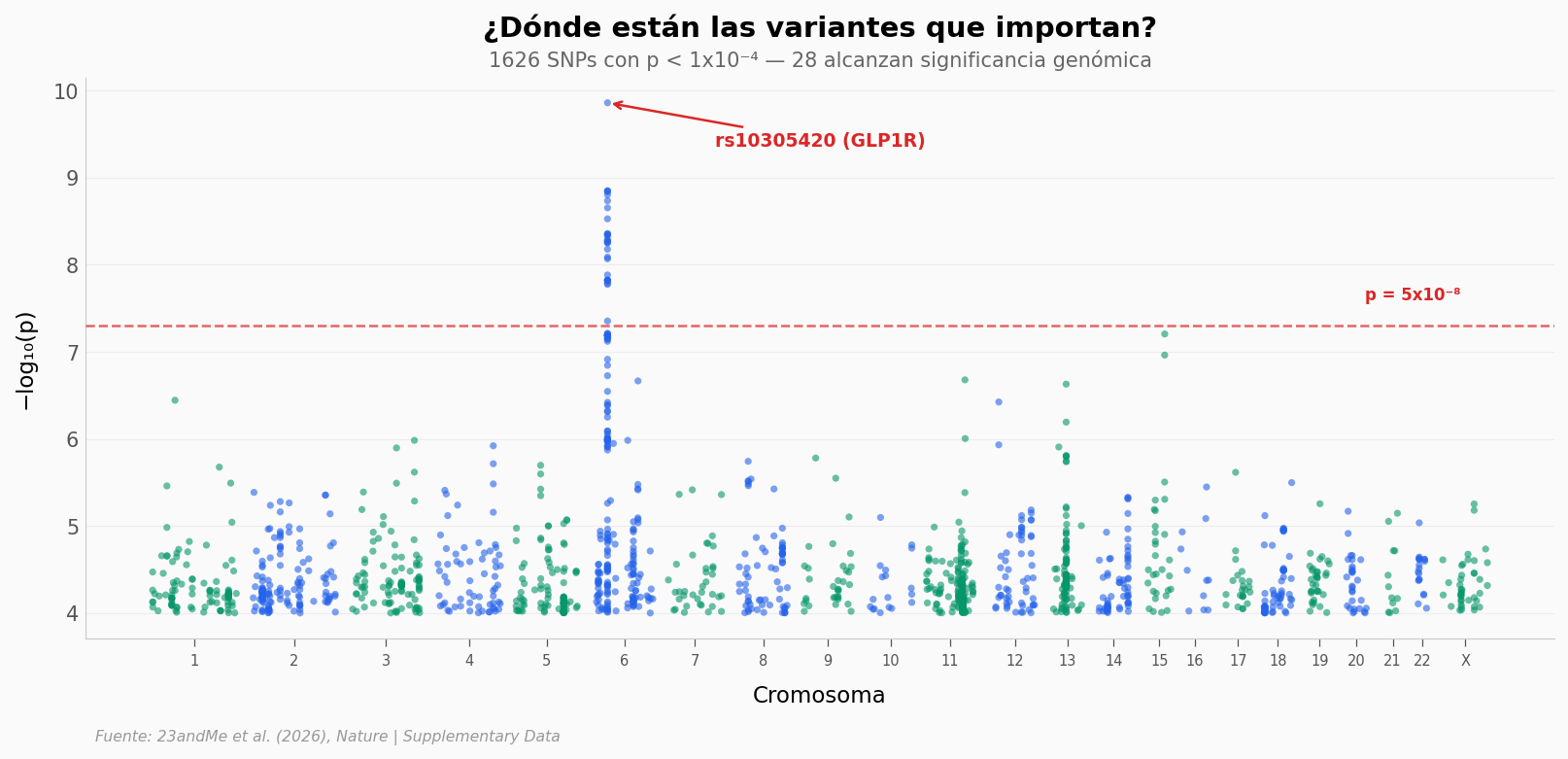
Lo que los datos soportan#
Afirmación |
¿Soportada? |
Detalle |
|---|---|---|
Variante rs10305420 se asocia con mayor pérdida de peso |
✅ |
Efecto = -0,64% IMC por alelo, p = 2,9 x 10 a la -10. El paper reporta -0,76 kg (conversión del modelo ajustado) |
Genotipo CC triplica riesgo de vómitos en tirzepatida |
✅ |
CC 11,8% vs GG 3,9% = 3,0x. Chi-cuadrado = 44,5, p = 2,2 x 10 a la -10 |
El efecto está restringido a tirzepatida |
✅ |
Semaglutida: CC 9,4% vs GG 9,7%, chi-cuadrado = 0,7, p = 0,70 |
Genética mejora predicción de efectos secundarios |
✅ |
AUC vómitos: +0,022 (0,679 vs 0,657). Mejora modesta pero consistente |
Genética mejora predicción de pérdida de peso |
⚠️ |
R cuadrado = 0,243 vs 0,242 (delta = +0,001). Mejora marginal. Dosis y tipo de fármaco dominan |
Medicina de precisión viable para obesidad |
⚠️ |
Los datos identifican variantes relevantes, pero el poder predictivo actual es limitado. El paper dice que estos resultados «sientan las bases» |
Limitaciones: Datos de encuesta auto-reportados (no mediciones clínicas de peso), muestra predominantemente europea (76%), rango limitado de dosis y duración. Los efectos son asociaciones (GWAS), no relaciones causales. El GWAS explica una fracción pequeña de la variabilidad total.
Ahora tú#
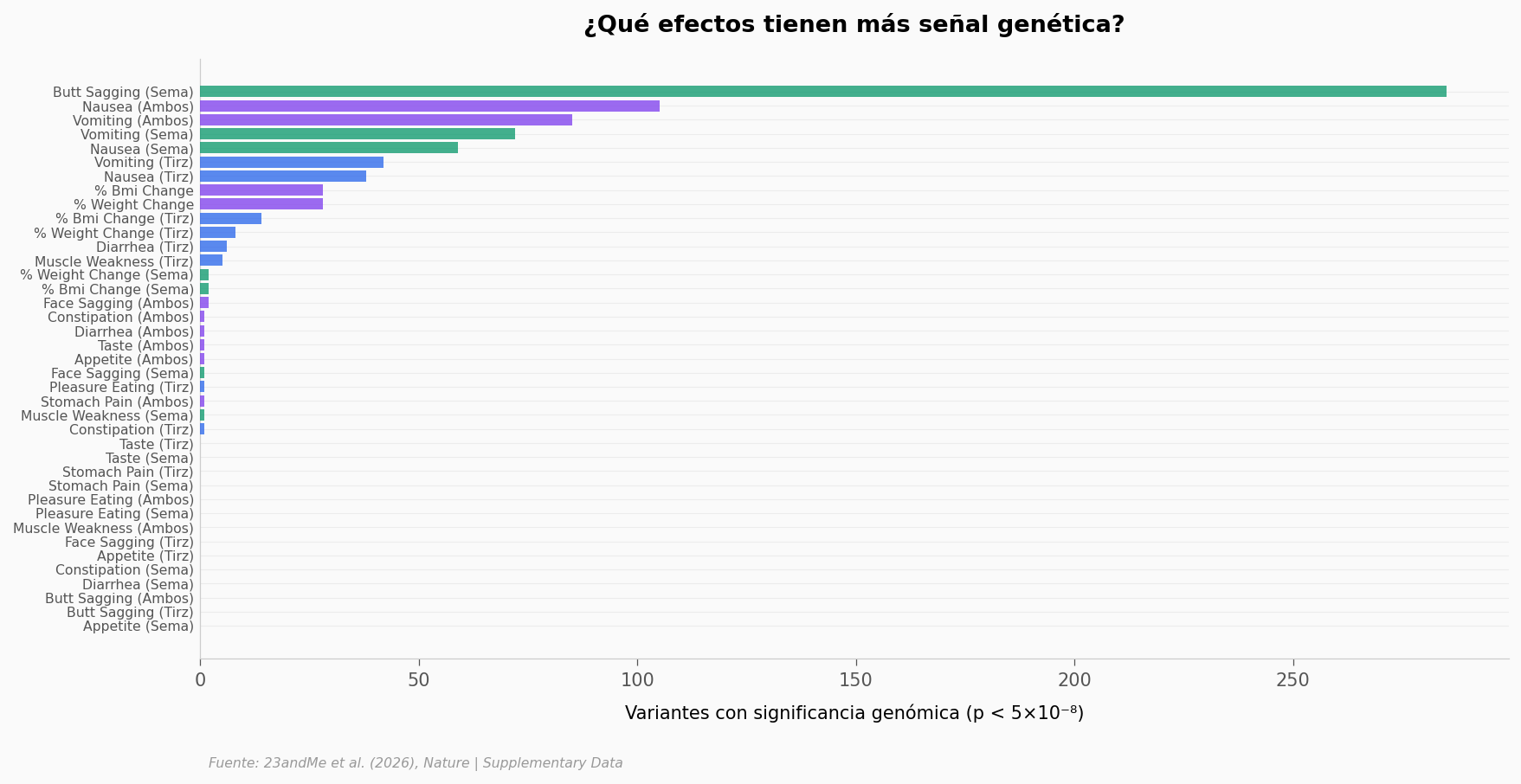
¿Cuántos SNPs alcanzan significancia genómica por fenotipo? Los datos incluyen 39 fenotipos (efectos secundarios + eficacia). Prueba a cargar y graficar el número de variantes significativas por fenotipo. ¿Cuáles efectos secundarios tienen más señal genética?
¿Hay diferencia por sexo en la prevalencia de efectos secundarios? Los datos de incluyen efectos secundarios desglosados por sexo. ¿Los hombres y las mujeres reportan las mismas tasas de náusea y vómitos?
¿La educación realmente importa? El modelo incluye «Years of Education» como predictor (coef = +0,29, p = 0,0002). ¿Por qué aparecería la educación en un modelo de pérdida de peso? Prueba a visualizar los coeficientes de las interacciones (Dosis x Tipo, Dosis x Días) y piensa en qué variables podrían ser confusoras.
# --- EXPERIMENTA AQUI ---
# Cuales efectos secundarios tienen mas senal genetica?
fig, ax = plt.subplots(figsize=(13, 6))
gf = gwas_fenotipos.sort_values('n_snps_gw_sig', ascending=True)
labels = []
for p in gf['phenotype']:
label = p.replace('glp_', '').replace('side_effect_', '')
label = label.replace('prct_', '% ')
label = label.replace('_semaglutide_or_tirzepatide', ' (ambos)')
label = label.replace('_semaglutide', ' (sema)')
label = label.replace('_tirzepatide', ' (tirz)')
label = label.replace('_', ' ').title()
labels.append(label)
y = np.arange(len(labels))
colors_bar = [COLOR_TIRZ if 'tirz' in l.lower() else
COLOR_SEMA if 'sema' in l.lower() else
COLOR_VIOLETA for l in labels]
ax.barh(y, gf['n_snps_gw_sig'].values, color=colors_bar, alpha=0.75, zorder=5)
ax.set_yticks(y)
ax.set_yticklabels(labels, fontsize=7.5)
ax.set_xlabel('Variantes con significancia genómica (p < 5×10\u207b\u2078)', fontsize=10)
ax.set_title('¿Qué efectos tienen más señal genética?', fontsize=13, fontweight='bold', pad=15)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/senales_por_fenotipo.png', dpi=200, bbox_inches='tight')
plt.show()
Paper: Genetic predictors of GLP1 receptor agonist weight loss and side effects DOI: 10.1038/s41586-026-10330-z Journal: Nature (2026) Datos: Supplementary Data (Supplementary Tables + GWAS Summary Statistics) Licencia datos: CC BY 4.0 (Nature Open Access) Notebook: Ciencia a Mordiscos — El Lab