
382 isoleucinas que sobraban#
¿Y si pudieras vivir con un aminoácido menos? Todos los organismos conocidos usan 20 aminoácidos canónicos para construir sus proteínas. El Wang Lab de Columbia decidió probar si una Escherichia coli podía funcionar con 19 — quitándole la isoleucina. Para empezar, tenían que rediseñar el ribosoma: identificaron y reemplazaron 382 residuos de Ile distribuidos en 50 proteínas ribosomales.
En este notebook recreamos ese conteo desde el FASTA público y exploramos cómo se distribuye Ile en el ribosoma — el dato que define el alcance del rediseño.
Paper: Toward life with a 19–amino acid alphabet through generative artificial intelligence design · Science (2026)
DOI: 10.1126/science.aeb5171
Datos: github.com/wanglabcumc/Ec19 · data/ecoli_ribosomal_genes.fasta
![]()
El ribosoma como punto de partida#
Para construir una bacteria sin isoleucina hay que reescribir todas sus proteínas esenciales sin ese aminoácido. El ribosoma — la maquinaria que fabrica proteínas — es el corazón del problema: si el ribosoma falla, la célula entera falla.
El equipo se centró en las 50 proteínas que forman el ribosoma de E. coli. Tomaron las secuencias naturales (wild-type), contaron cuánta isoleucina contenían, y usaron modelos generativos de IA (basados en lenguaje de proteínas y estructura) para rediseñar versiones sin Ile que siguieran plegándose y funcionando.
Aquí miramos solo lo primero: la materia prima del rediseño.
# ══════════════════════════════════════════════════════════════
# Configuración — modifica estos valores para explorar
# ══════════════════════════════════════════════════════════════
PROT_DESTACADA = 'rpsA' # proteína a destacar en gráficas (la del outlier)
PROT_CONCENTRACION = 10 # top-N para "concentración de trabajo de rediseño"
FUENTE = 'Fuente: paper Science (2026), DOI 10.1126/science.aeb5171 | Datos: github.com/wanglabcumc/Ec19'
COLOR_DATOS = '#2563EB' # azul CaM
COLOR_ALERTA = '#DC2626' # rojo (outlier / Ile destacada)
COLOR_REFERENCIA = '#D97706' # amber (mediana / referencia)
COLOR_SECUNDARIO = '#059669' # emerald (datos comparativos)
COLOR_CONTEXTO = '#BBBBBB' # gris (background)
# ── Imports ──
import os, urllib.request
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
# Estilo CaM (local → GitHub raw fallback)
style_file = '../../cam.mplstyle'
if not os.path.exists(style_file):
style_file = '/tmp/cam.mplstyle'
if not os.path.exists(style_file):
urllib.request.urlretrieve(
'https://raw.githubusercontent.com/Ciencia-a-Mordiscos/lab/main/cam.mplstyle',
style_file)
plt.style.use(style_file)
# ── Carga de datos ──
df_genes = pd.read_csv('datos/ribosomal_genes_ile.csv')
df_aa = pd.read_csv('datos/aa_composition_global.csv')
# ── Resumen ──
n_prot = len(df_genes)
n_ile_total = int(df_genes['ile_count'].sum())
n_residuos = int(df_genes['length_aa'].sum())
pct_ile_global = n_ile_total / n_residuos * 100
print(f'Proteínas ribosomales analizadas: {n_prot}')
print(f'Residuos totales: {n_residuos:,}'.replace(',', '.'))
print(f'Isoleucinas totales: {n_ile_total} ({pct_ile_global:.2f} % del total)')
print(f'Cross-check vs paper: el paper reporta 382 Ile → coincidencia exacta.')
Proteínas ribosomales analizadas: 50
Residuos totales: 6.815
Isoleucinas totales: 382 (5.61 % del total)
Cross-check vs paper: el paper reporta 382 Ile → coincidencia exacta.
Cómo se reparten las 382 isoleucinas#
Aquí está.
fig, ax = plt.subplots(figsize=(13, 5.5))
ile_counts = df_genes['ile_count'].values
median = np.median(ile_counts)
mean = ile_counts.mean()
# Histograma
n, bins, patches = ax.hist(ile_counts, bins=np.arange(0, 33, 1),
color=COLOR_DATOS, alpha=0.55,
edgecolor=COLOR_DATOS, linewidth=0.8)
# Línea de mediana
ax.axvline(median, color=COLOR_REFERENCIA, linewidth=1.5, linestyle='--', alpha=0.85)
ax.annotate(f'Mediana = {median:.0f} Ile',
xy=(median, n.max()*0.85), xytext=(median+3, n.max()*0.95),
fontsize=10, fontweight='bold', color=COLOR_REFERENCIA,
arrowprops=dict(arrowstyle='->', color=COLOR_REFERENCIA, lw=1.2))
# Outlier rpsA
outlier_row = df_genes.loc[df_genes['ile_count'].idxmax()]
ax.annotate(f"{outlier_row['gene_name']}: {int(outlier_row['ile_count'])} Ile\n(557 aa, la más larga)",
xy=(outlier_row['ile_count'], 1), xytext=(22, n.max()*0.5),
fontsize=10, fontweight='bold', color=COLOR_ALERTA,
arrowprops=dict(arrowstyle='->', color=COLOR_ALERTA, lw=1.5))
ax.set_xlabel('Isoleucinas por proteína')
ax.set_ylabel('Número de proteínas')
ax.set_title('¿Cuántas Ile hay que reemplazar en cada pieza del ribosoma?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03, f'{n_prot} proteínas ribosomales · {n_ile_total} Ile en total',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.set_xlim(-0.5, 32)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/distribucion_ile_por_proteina.png', dpi=200, bbox_inches='tight')
plt.show()
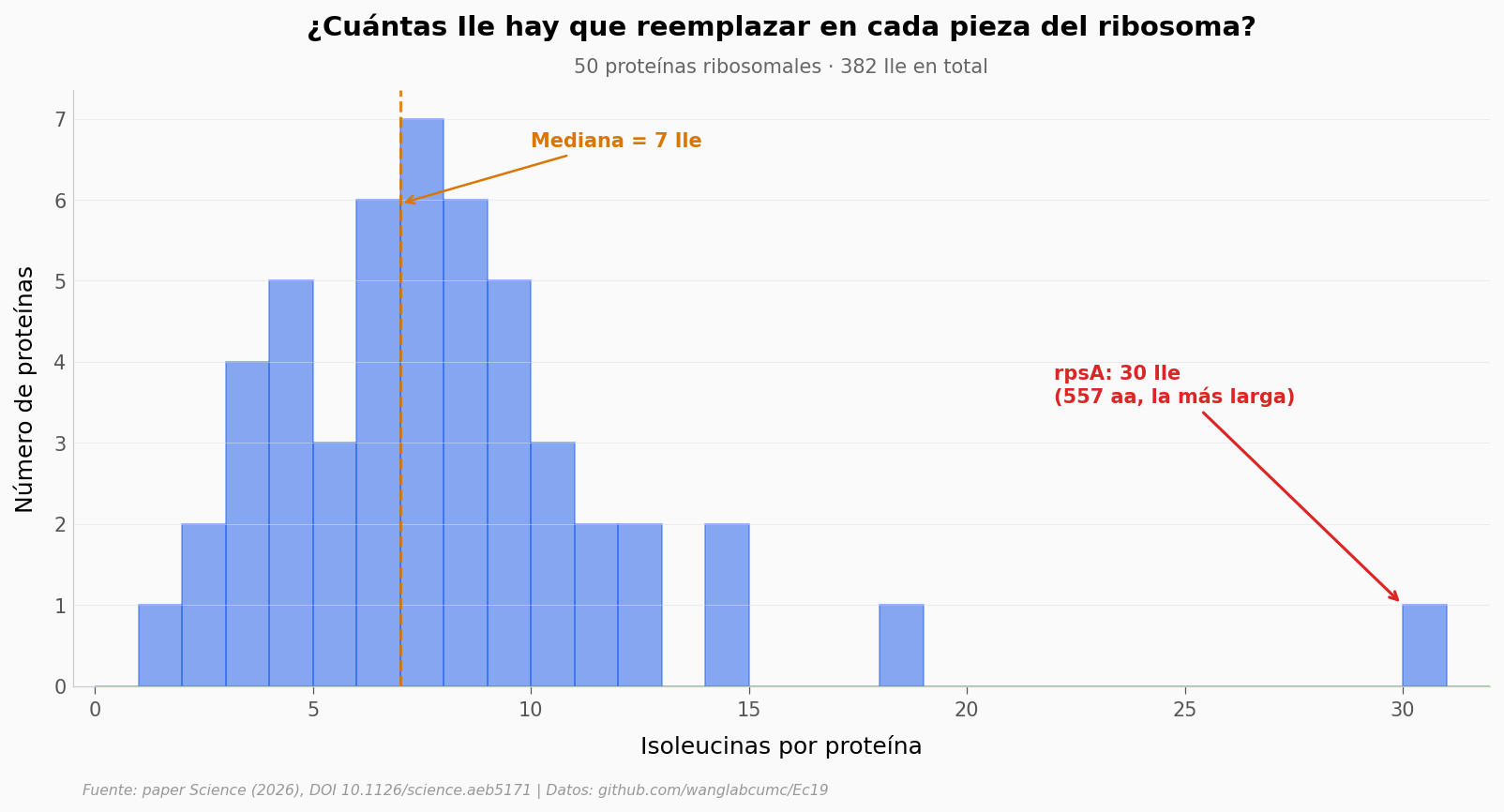
La distribución se inclina fuertemente hacia la izquierda: la mediana es 7 isoleucinas por proteína y la mayoría se concentra entre 4 y 9. Pero hay un outlier extremo: una proteína sola (rpsA, la subunidad más larga del ribosoma) acumula 30 Ile — cuatro veces el promedio. Removerle la isoleucina a rpsA es, por sí mismo, casi el 8 % del trabajo total del rediseño.
La asimetría importa: si Ile estuviera repartido uniformemente, bastaría una receta general. Como no lo está, cada proteína requiere su propio plan.
Las 10 proteínas que concentran el rediseño#
¿En cuántas proteínas se concentra realmente el trabajo? Veamos las que más Ile tienen.
top10 = df_genes.nlargest(PROT_CONCENTRACION, 'ile_count').sort_values('ile_count')
top10_sum = int(top10['ile_count'].sum())
top10_pct = top10_sum / n_ile_total * 100
fig, ax = plt.subplots(figsize=(13, 5.5))
colors = [COLOR_ALERTA if g == PROT_DESTACADA else COLOR_DATOS for g in top10['gene_name']]
bars = ax.barh(top10['gene_name'], top10['ile_count'], color=colors,
alpha=0.85, edgecolor='white', linewidth=0.8)
# Etiquetas con conteo + porcentaje de la proteína
for bar, count, pct in zip(bars, top10['ile_count'], top10['ile_pct']):
ax.text(bar.get_width() + 0.4, bar.get_y() + bar.get_height()/2,
f'{int(count)} ({pct:.1f} %)',
va='center', fontsize=9, color='#444444')
ax.set_xlabel('Isoleucinas por proteína')
ax.set_title(f'Top {PROT_CONCENTRACION}: concentran {top10_sum} de {n_ile_total} Ile ({top10_pct:.1f} %)',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03,
f'El rediseño no se reparte parejo — 10 proteínas acumulan más de un tercio del trabajo',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.set_xlim(0, 33)
# Etiqueta inline sobre rpsA
rpsA_idx = list(top10['gene_name']).index(PROT_DESTACADA)
ax.text(31, rpsA_idx, 'la más larga\n(557 aa)', va='center', ha='left',
fontsize=8, color=COLOR_ALERTA, fontweight='bold', style='italic')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/top10_ile_por_proteina.png', dpi=200, bbox_inches='tight')
plt.show()
print(f'\nTop {PROT_CONCENTRACION} concentra {top10_sum}/{n_ile_total} Ile = {top10_pct:.1f} %')
print(f'rpsA sola: {int(outlier_row["ile_count"])} Ile = {outlier_row["ile_count"]/n_ile_total*100:.1f} % del rediseño')
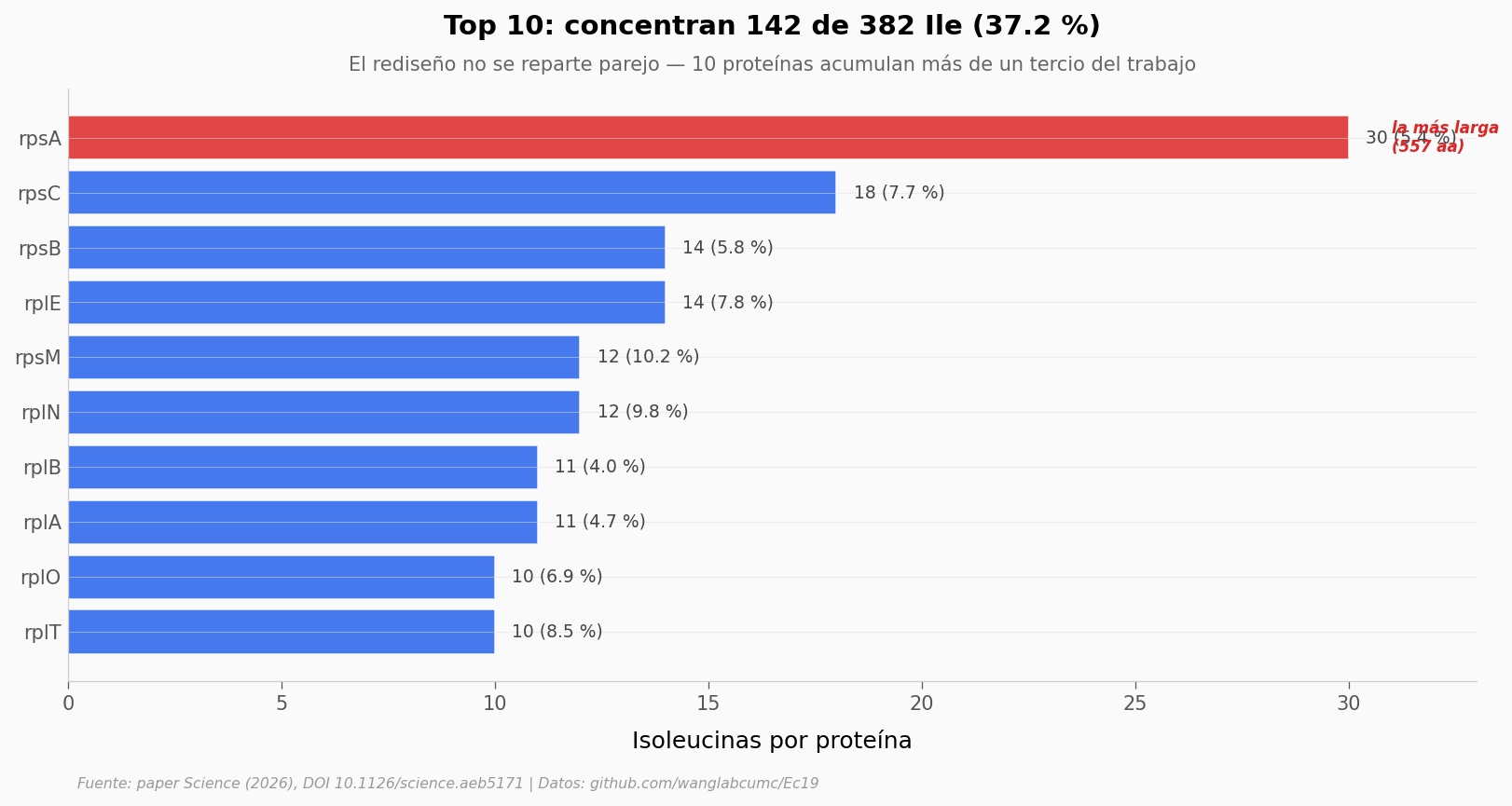
Top 10 concentra 142/382 Ile = 37.2 %
rpsA sola: 30 Ile = 7.9 % del rediseño
¿Es Ile especialmente abundante en el ribosoma?#
Si Ile fuera uno de los aminoácidos más usados, su eliminación sería desproporcionadamente difícil. Pero si es minoritario, ¿por qué se eligió a Ile y no a otro? Veamos cómo se compara con los demás aminoácidos en estas mismas 50 proteínas.
# Normalizar nombre de columna (CSV usa pct_of_total)
df_aa = df_aa.rename(columns={'pct_of_total': 'pct'})
# Ordenar por count asc para que barh muestre el más abundante arriba
df_aa_sorted = df_aa.sort_values('count', ascending=True).reset_index(drop=True)
df_aa_sorted['rank'] = df_aa_sorted['count'].rank(ascending=False).astype(int)
# Diccionario aa → nombre completo
aa_names = {
'A': 'Ala', 'R': 'Arg', 'N': 'Asn', 'D': 'Asp', 'C': 'Cys',
'E': 'Glu', 'Q': 'Gln', 'G': 'Gly', 'H': 'His', 'I': 'Ile',
'L': 'Leu', 'K': 'Lys', 'M': 'Met', 'F': 'Phe', 'P': 'Pro',
'S': 'Ser', 'T': 'Thr', 'W': 'Trp', 'Y': 'Tyr', 'V': 'Val'
}
df_aa_sorted['label'] = df_aa_sorted['amino_acid'].map(lambda a: f'{aa_names[a]} ({a})')
fig, ax = plt.subplots(figsize=(13, 6.5))
colors = [COLOR_ALERTA if a == 'I' else COLOR_DATOS for a in df_aa_sorted['amino_acid']]
bars = ax.barh(df_aa_sorted['label'], df_aa_sorted['pct'], color=colors,
alpha=0.85, edgecolor='white', linewidth=0.8)
for bar, pct in zip(bars, df_aa_sorted['pct']):
ax.text(bar.get_width() + 0.12, bar.get_y() + bar.get_height()/2,
f'{pct:.2f} %', va='center', fontsize=8.5, color='#555555')
# Anotación sobre Ile
ile_pos = list(df_aa_sorted['amino_acid']).index('I')
ile_rank = int(df_aa_sorted.loc[ile_pos, 'rank'])
ax.annotate(f'Ile: 8º de 20 ({pct_ile_global:.2f} %) — ni el más usado ni de los más raros',
xy=(df_aa_sorted.loc[ile_pos, 'pct'], ile_pos),
xytext=(7.5, ile_pos - 5.5),
fontsize=10, fontweight='bold', color=COLOR_ALERTA,
arrowprops=dict(arrowstyle='->', color=COLOR_ALERTA, lw=1.5))
ax.set_xlabel('Porcentaje del total de residuos')
ax.set_title('¿Dónde encaja la isoleucina entre los 20 aminoácidos?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03, f'Composición global en {n_prot} proteínas ribosomales',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.set_xlim(0, 13)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/composicion_global_aa.png', dpi=200, bbox_inches='tight')
plt.show()
¿Tan extremo es este alfabeto?#
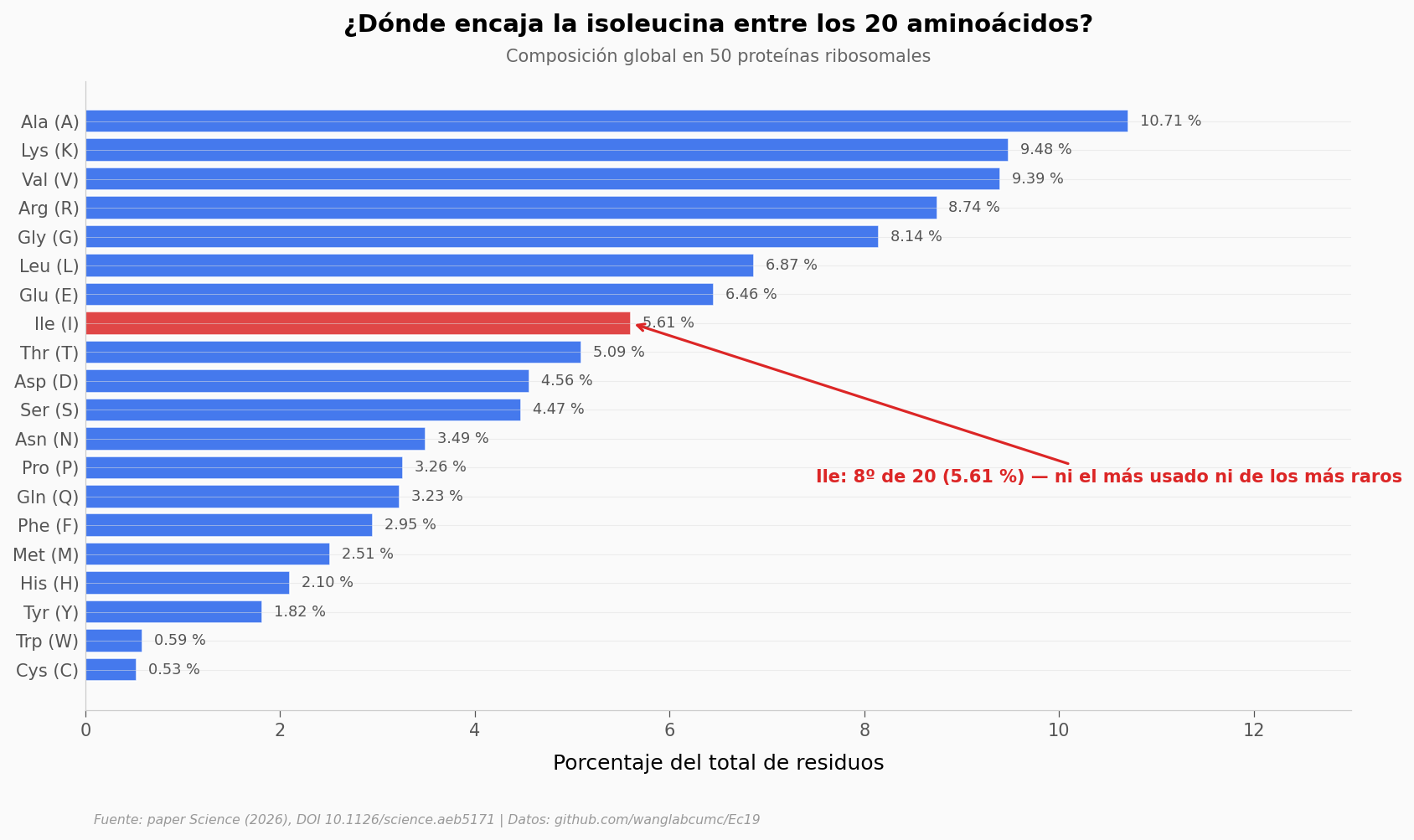
Ile no es el aminoácido más abundante (lo es Ala, con 10,71 %) ni de los más raros (Trp está en el último puesto con menos del 1,5 %). Está en mitad de la tabla: el octavo de veinte, con 5,61 % de los residuos. Suficientemente común para estar en casi todas las proteínas ribosomales — pero no tanto como para hacer el rediseño imposible.
¿Cómo se ve la concentración de Ile por proteína? Algunas tienen mucha más Ile de lo esperado para una proteína «promedio».
fig, ax = plt.subplots(figsize=(10, 5))
ile_pcts = df_genes['ile_pct'].values
n, bins, patches = ax.hist(ile_pcts, bins=20, color=COLOR_DATOS, alpha=0.4,
edgecolor=COLOR_DATOS, linewidth=0.8)
y_max = n.max() * 1.25
ax.set_ylim(0, y_max)
# Línea: promedio global (5.61%)
ax.axvline(pct_ile_global, color=COLOR_REFERENCIA, linewidth=2.0, linestyle='--')
ax.text(pct_ile_global + 0.15, y_max*0.92,
f'Global: {pct_ile_global:.2f} %', fontsize=10, color=COLOR_REFERENCIA,
fontweight='bold')
# Línea: máximo por proteína (10.17% rpsM/rpmD)
max_pct = ile_pcts.max()
ax.axvline(max_pct, color=COLOR_ALERTA, linewidth=2.0)
ax.text(max_pct + 0.15, y_max*0.78,
f'Máximo por proteína:\n{max_pct:.2f} % (rpsM, rpmD)',
fontsize=10, color=COLOR_ALERTA, fontweight='bold')
# Flecha bidireccional global ↔ máximo
ax.annotate('', xy=(max_pct, y_max*0.55), xytext=(pct_ile_global, y_max*0.55),
arrowprops=dict(arrowstyle='<->', color='#666666', lw=1.5))
ax.text((pct_ile_global + max_pct)/2, y_max*0.6,
f'{(max_pct - pct_ile_global):.1f} puntos porcentuales',
ha='center', fontsize=9, color='#666666', style='italic')
ax.set_xlabel('% Ile en la proteína')
ax.set_ylabel('Número de proteínas')
ax.set_title('¿Qué tan concentrada puede estar la isoleucina en una sola proteína?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03,
f'rpsM y rpmD casi duplican el promedio global del ribosoma',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/histograma_pct_ile.png', dpi=200, bbox_inches='tight')
plt.show()
print(f'Promedio global de Ile: {pct_ile_global:.2f} %')
print(f'Proteína con mayor % Ile: {max_pct:.2f} % (casi 2× el promedio)')
print(f'Proteínas con %Ile por encima del promedio: {(df_genes["ile_pct"] > pct_ile_global).sum()} de {n_prot}')
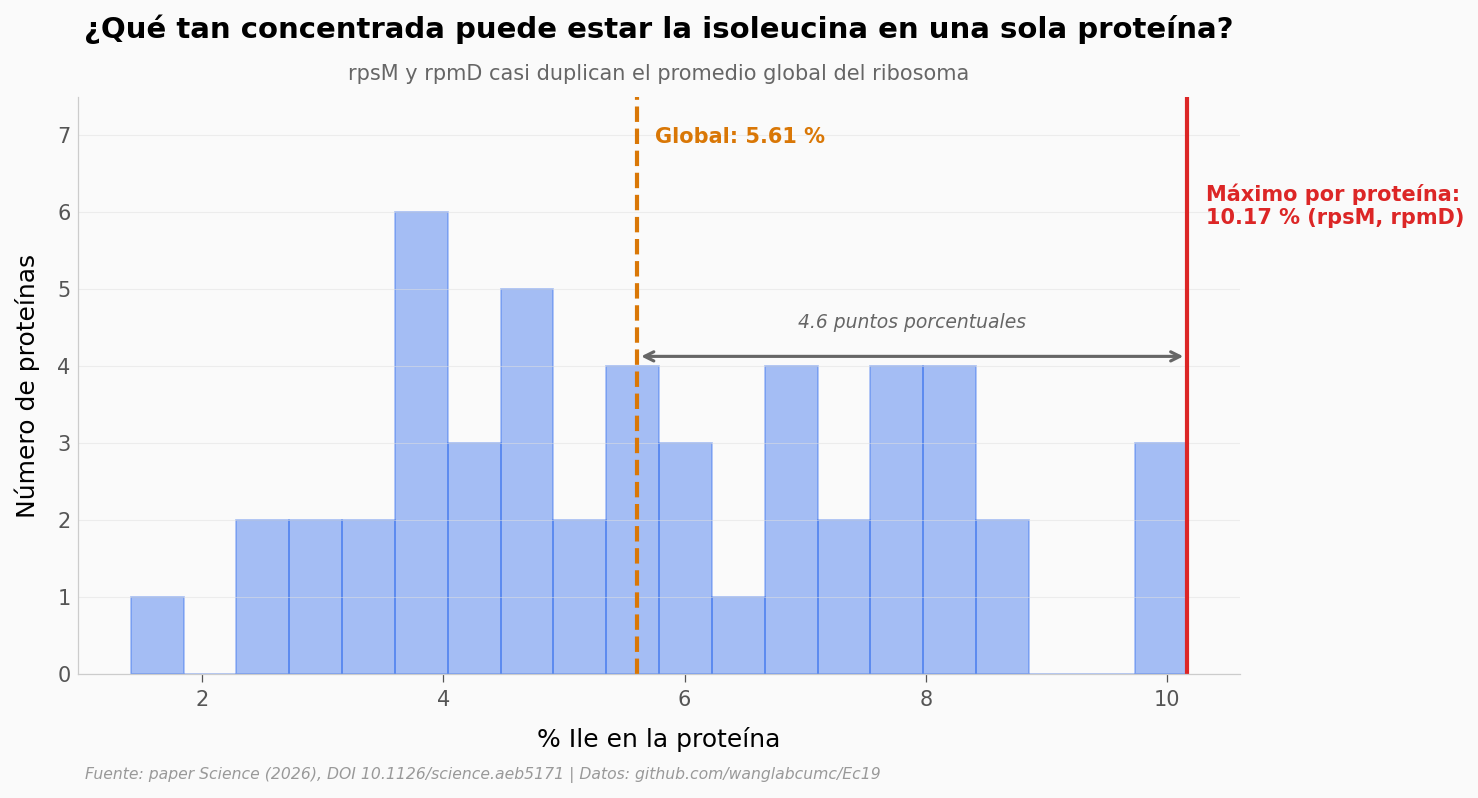
Promedio global de Ile: 5.61 %
Proteína con mayor % Ile: 10.17 % (casi 2× el promedio)
Proteínas con %Ile por encima del promedio: 25 de 50
Lo que los datos soportan#
Afirmación |
¿Soportada? |
Detalle |
|---|---|---|
El ribosoma de E. coli tiene 382 residuos de Ile distribuidos en 50 proteínas |
✅ |
Recuento directo del FASTA público coincide exactamente con el headline del paper (diferencia = 0). |
La distribución de Ile por proteína es asimétrica con outlier extremo ( |
✅ |
min=1, mediana=7, max=30. |
Las 10 proteínas con más Ile concentran 142/382 = 37 % del trabajo de rediseño |
✅ |
Cálculo directo sobre el FASTA. |
Ile es el 8º aminoácido más usado en el ribosoma (5,61 % del total) |
✅ |
Conteo global sobre 6.815 residuos. Por debajo de Ala, Lys, Val, Arg, Gly, Leu, Glu. |
El equipo reemplazó sistemáticamente los 382 Ile y produjo una E. coli viable y evolutivamente estable sin Ile en el ribosoma |
✅ |
Afirmación directa del paper («We systematically replaced… to produce a viable, evolutionarily stable cell»). Verificación experimental del laboratorio, no de este notebook. |
Modelos de lenguaje proteico y de estructura fueron necesarios para diseñar las variantes funcionales sin Ile |
✅ |
El paper lo declara como necesidad técnica («PLMs and structure-based models were necessary»). |
Este trabajo crea un organismo completo de 19 aminoácidos |
⚠️ |
Matiz importante: el paper rediseña el ribosoma sin Ile y demuestra una célula viable con esos componentes. Reemplazar Ile en TODO el proteoma de E. coli queda como trabajo futuro — el propio paper lo enmarca como roadmap. |
Limitaciones: El notebook explora la composición de Ile antes del rediseño. No verifica la viabilidad de las variantes diseñadas (eso requiere experimentos de laboratorio reportados en el paper). El FASTA pública del repo incluye únicamente las 50 secuencias wild-type — no las versiones rediseñadas ni los datos de crecimiento.
Ahora tú#
Tres preguntas para explorar con los datos:
¿Y si en vez de Ile hubieran quitado Val? Val (valina) es químicamente parecida a Ile — ambas son hidrofóbicas y ramificadas. ¿Cuántos residuos habría que reemplazar?
¿Es la longitud lo que predice cuántas Ile tendrá una proteína? Si lo es, no hay nada «extraordinario» en
rpsA. Calcula la correlación entre longitud y conteo de Ile.¿Hay proteínas que apenas tengan Ile? Mira el extremo opuesto: las que el rediseño casi no tocó.
La celda siguiente responde la pregunta 1 — modifica el código para responder las otras.
# --- EXPERIMENTA AQUÍ ---
# Pregunta 1: ¿Cuánto trabajo habría sido quitar Val en vez de Ile?
n_val_total = int(df_genes['val_count'].sum())
pct_val_global = n_val_total / n_residuos * 100
print(f'Si hubieran elegido Val en vez de Ile:')
print(f' - Residuos a reemplazar: {n_val_total} (vs {n_ile_total} de Ile)')
print(f' - Porcentaje global: {pct_val_global:.2f} % (vs {pct_ile_global:.2f} % de Ile)')
print(f' - Trabajo extra: {n_val_total - n_ile_total} residuos más ({(n_val_total/n_ile_total - 1)*100:.0f} %)')
print()
print('Val es más abundante en el ribosoma → un alfabeto sin Val sería un rediseño más grande.')
print('Ile probablemente se eligió por una combinación de ser dispensable y minimizar el alcance.')
# Pista para pregunta 2:
# from scipy import stats
# r, p = stats.spearmanr(df_genes['length_aa'], df_genes['ile_count'])
# print(f'\nCorrelación longitud ↔ Ile: ρ = {r:.3f}, p = {p:.4f}')
# Pista para pregunta 3:
# print(df_genes.nsmallest(5, 'ile_count')[['gene_name', 'length_aa', 'ile_count', 'ile_pct']])
Si hubieran elegido Val en vez de Ile:
- Residuos a reemplazar: 640 (vs 382 de Ile)
- Porcentaje global: 9.39 % (vs 5.61 % de Ile)
- Trabajo extra: 258 residuos más (68 %)
Val es más abundante en el ribosoma → un alfabeto sin Val sería un rediseño más grande.
Ile probablemente se eligió por una combinación de ser dispensable y minimizar el alcance.
Fuentes#
Paper: Toward life with a 19–amino acid alphabet through generative artificial intelligence design
Science, 2026-04-30 · paywall
Dataset canónico: Code accompanying the paper — data/ecoli_ribosomal_genes.fasta (50 E. coli ribosomal gene wild-type sequences)
GitHub (wanglabcumc/Ec19) — secuencias FASTA wild-type del ribosoma
Datos del pipeline computacional: Zenodo · Zenodo
15 afirmaciones del notebook verificadas contra estas fuentes
Repo: github.com/Ciencia-a-Mordiscos/lab · Notebook reproducible — todos los datos provienen del FASTA público del Wang Lab.