
Vida que se delata por cómo se reparte#
Si encontráramos un meteorito con 27 tipos de aminoácidos, ¿diríamos que hay vida? Bennu — la roca traída de regreso por la sonda OSIRIS-REx en 2023 — tiene exactamente eso. Y, aún así, es abiótica.
Un nuevo paper en Nature Astronomy propone una bisagra: la vida no se distingue por cuántos tipos de moléculas hay, sino por cómo se reparten. Aquí abrimos sus datos para verlo con nuestros ojos.
Paper: Molecular diversity as a biosignature · Yoffe et al., Nature Astronomy (2026-05-11) Datos: Zenodo — abundancias y errores Video: [Pendiente]
![]()
La pregunta que aterriza esto#
Las misiones planetarias pueden medir abundancias relativas de moléculas — eso es accesible. Lo que no pueden hacer fácil es medir quiralidad o composición isotópica con la precisión que la biología demanda en otro mundo. El equipo de Yoffe et al. junta 69 muestras de aminoácidos: 30 abióticas (meteoritos, simulaciones, química prebiótica), 28 bióticas (microbios, fósiles antiguos, suelos), y 11 mixtas (donde no se puede separar la fracción viva).
Sobre cada muestra calculan la entropía de Shannon — una medida que sube cuando las moléculas se reparten parejo, y baja cuando una o dos dominan. Su tesis: la vida tiende a repartir, la química abiótica tiende a concentrarse. Veámoslo.
# ══════════════════════════════════════════════════════════════
# Configuración — modifica estos valores para explorar
# ══════════════════════════════════════════════════════════════
COLOR_ABIOTIC = '#D97706' # Ámbar — química sin vida
COLOR_BIOTIC = '#059669' # Esmeralda — vida confirmada
COLOR_MIXED = '#7C3AED' # Violeta — origen ambiguo
COLOR_ALERTA = '#DC2626'
COLOR_DATOS = '#2563EB'
FUENTE = 'Fuente: Yoffe et al. (2026), Nature Astronomy | Datos: Zenodo 16566388 (CC-BY-4.0)'
# Setup
import os, sys, urllib.request, warnings
warnings.filterwarnings('ignore')
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from scipy import stats
# Estilo CaM (local en el repo o fallback a raw GitHub)
style_file = '../../cam.mplstyle'
if not os.path.exists(style_file):
style_file = '/tmp/cam.mplstyle'
if not os.path.exists(style_file):
urllib.request.urlretrieve(
'https://raw.githubusercontent.com/Ciencia-a-Mordiscos/lab/main/cam.mplstyle',
style_file,
)
plt.style.use(style_file)
# Carga de datos
aa = pd.read_csv('datos/AA_diversity_per_sample.csv')
fa = pd.read_csv('datos/FA_diversity_per_sample.csv')
aa_ab = pd.read_csv('datos/AA_abundances.csv')
print(f'Aminoácidos: {len(aa)} muestras')
print(aa['class_label'].value_counts().to_string())
print(f'\nÁcidos grasos: {len(fa)} muestras')
print(fa['class_label'].value_counts().to_string())
Aminoácidos: 69 muestras
class_label
abiotic 30
biotic 28
mixed 11
Ácidos grasos: 34 muestras
class_label
biotic 18
abiotic 16
Aquí está.#
Cada punto es una muestra. El eje vertical es la diversidad de Shannon de sus aminoácidos. Las medias y errores son media ± SEM por clase.
fig, ax = plt.subplots(figsize=(11, 5.5))
groups = [
('Abiótico', aa[aa['class_label']=='abiotic']['H'].values, COLOR_ABIOTIC, 0),
('Mixto', aa[aa['class_label']=='mixed']['H'].values, COLOR_MIXED, 1.4),
('Biótico', aa[aa['class_label']=='biotic']['H'].values, COLOR_BIOTIC, 2.8),
]
np.random.seed(42)
for label, vals, color, pos in groups:
n = len(vals)
x_jitter = np.linspace(pos - 0.18, pos + 0.18, n)
np.random.shuffle(x_jitter)
ax.scatter(x_jitter, vals, color=color, s=42, alpha=0.62,
edgecolors='white', linewidths=0.5, zorder=5)
mean = vals.mean()
sem = vals.std(ddof=1) / np.sqrt(n)
ax.errorbar(pos, mean, yerr=sem, fmt='_', color=color,
markersize=28, markeredgewidth=3,
capsize=8, capthick=1.6, zorder=6)
ax.text(pos, mean + sem + 0.18, f'H = {mean:.2f}',
ha='center', fontsize=10, fontweight='bold', color=color, zorder=7)
ax.set_xticks([0, 1.4, 2.8])
ax.set_xticklabels(
[f'Abiótico (n={len(groups[0][1])})',
f'Mixto (n={len(groups[1][1])})',
f'Biótico (n={len(groups[2][1])})'],
fontsize=10, fontweight='bold',
)
for tick, (_, _, color, _) in zip(ax.get_xticklabels(), groups):
tick.set_color(color)
ax.set_ylabel('Diversidad de Shannon (H, nats)', fontsize=11)
ax.set_ylim(0.8, 3.3)
ax.set_title('¿Qué tan pareja se reparte la mezcla?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03, 'Aminoácidos · 69 muestras en total',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.text(0.98, 0.02, '━ media ± SEM',
transform=ax.transAxes, fontsize=8, color='#999999',
ha='right', va='bottom', style='italic')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/diversidad_por_clase.png', dpi=200, bbox_inches='tight')
plt.show()
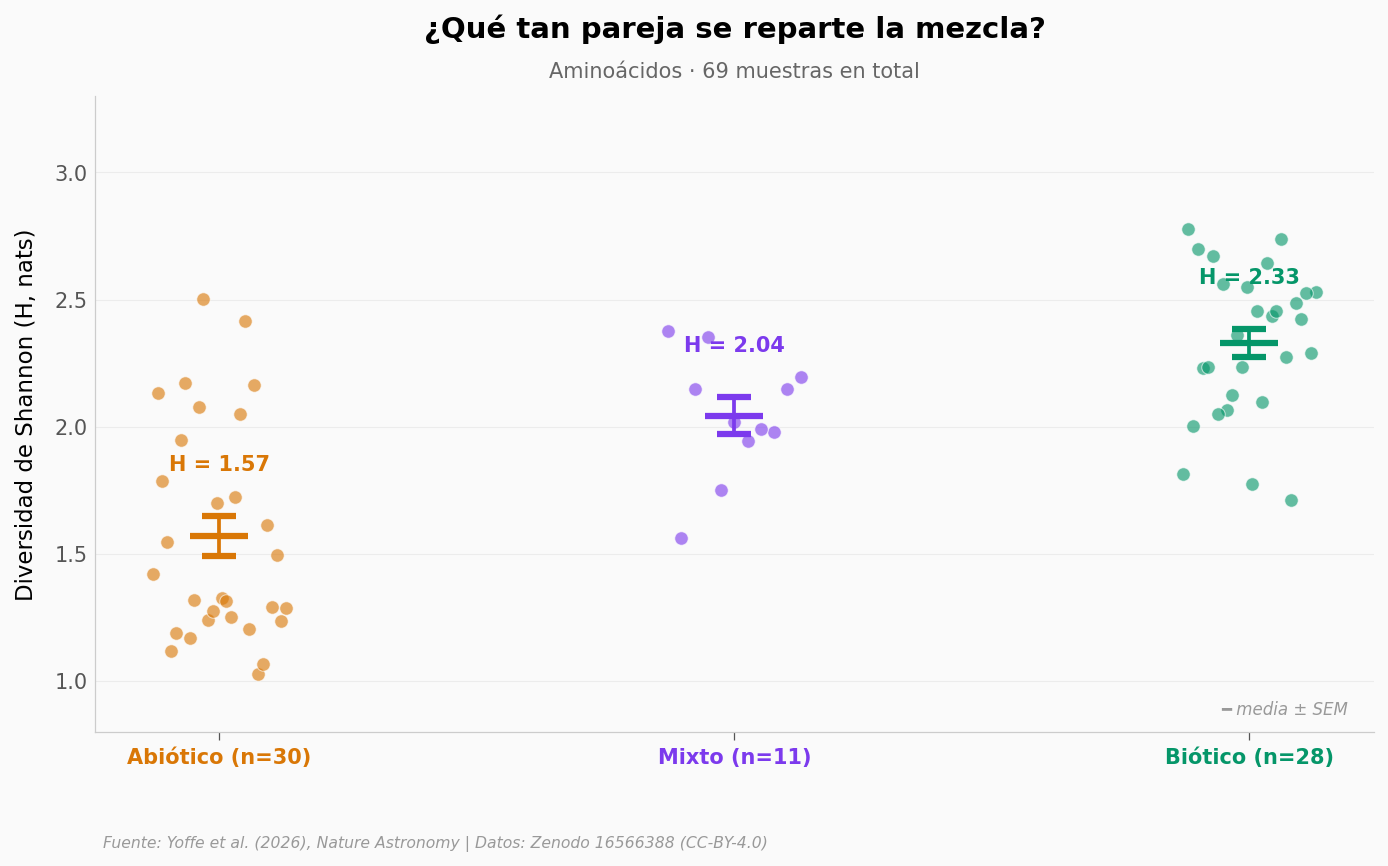
La distancia entre las nubes verde y ámbar es la pieza central. Las muestras bióticas se concentran arriba (mediana ≈ 2,39); las abióticas, abajo (mediana ≈ 1,37). El solapamiento existe — hay meteoritos como Murchison y Allende con diversidad alta —, pero la nube de la vida vive separada de la nube del polvo cósmico.
La diferencia, además de visible, es estadísticamente fuerte:
# Test Mann-Whitney U (one-sided: biotic > abiotic) — no asume normalidad
H_biotic = aa[aa['class_label']=='biotic']['H'].values
H_abiotic = aa[aa['class_label']=='abiotic']['H'].values
u_stat, p_val = stats.mannwhitneyu(H_biotic, H_abiotic, alternative='greater')
# Cohen's d (pooled) — tamaño del efecto
n1, n2 = len(H_biotic), len(H_abiotic)
s_pool = np.sqrt(((n1-1)*H_biotic.var(ddof=1) + (n2-1)*H_abiotic.var(ddof=1)) / (n1+n2-2))
cohen_d = (H_biotic.mean() - H_abiotic.mean()) / s_pool
print(f'Biótico: media H = {H_biotic.mean():.3f} (n={n1})')
print(f'Abiótico: media H = {H_abiotic.mean():.3f} (n={n2})')
print(f'Diferencia: {H_biotic.mean() - H_abiotic.mean():+.3f} nats '
f'({100*(H_biotic.mean() - H_abiotic.mean())/H_abiotic.mean():.1f}%)')
print(f'Mann-Whitney U = {u_stat:.0f}, p = {p_val:.2e}')
print(f"Cohen's d (pooled) = {cohen_d:.2f} (d>0,8 = efecto grande)")
Biótico: media H = 2.328 (n=28)
Abiótico: media H = 1.568 (n=30)
Diferencia: +0.760 nats (48.5%)
Mann-Whitney U = 766, p = 3.80e-08
Cohen's d (pooled) = 2.06 (d>0,8 = efecto grande)
La paradoja: lo abiótico tiene más tipos#
La intuición ingenua dice «más vida = más variedad química». Los datos dicen lo contrario: las muestras abióticas tienen, en promedio, más tipos distintos de aminoácidos que las bióticas (16,3 vs 14,1). ¿Cómo cuadra eso con que la vida sea «más diversa»?
La clave está en cómo se reparten esos tipos. Veamos.
# Calcular tipos presentes (n_present) por clase y plotear vs H
fig, axes = plt.subplots(1, 2, figsize=(13, 5.5))
# --- Panel A: barras de tipos promedio (richness) ---
ax = axes[0]
class_order = ['abiotic', 'mixed', 'biotic']
labels_es = ['Abiótico', 'Mixto', 'Biótico']
colors = [COLOR_ABIOTIC, COLOR_MIXED, COLOR_BIOTIC]
richness_means = [aa[aa['class_label']==c]['n_present'].mean() for c in class_order]
richness_sems = [aa[aa['class_label']==c]['n_present'].std(ddof=1)
/ np.sqrt(len(aa[aa['class_label']==c])) for c in class_order]
bars = ax.bar(labels_es, richness_means, yerr=richness_sems,
color=colors, alpha=0.75, edgecolor='white', linewidth=1.5,
capsize=6, error_kw={'ecolor': '#666666', 'lw': 1.2})
for bar, mean in zip(bars, richness_means):
ax.text(bar.get_x() + bar.get_width()/2, mean + 0.4, f'{mean:.1f}',
ha='center', fontsize=10, fontweight='bold')
ax.set_ylabel('Tipos de aminoácidos presentes (media)', fontsize=11)
ax.set_ylim(0, 22)
ax.set_title('Cuántos tipos hay (riqueza)', fontsize=13, fontweight='bold', pad=10)
# --- Panel B: relación tipos vs diversidad ---
ax = axes[1]
for c, label, color in zip(class_order, labels_es, colors):
sub = aa[aa['class_label']==c]
ax.scatter(sub['n_present'], sub['H'],
color=color, s=45, alpha=0.7,
edgecolors='white', linewidths=0.5,
label=label, zorder=5)
ax.set_xlabel('Tipos de aminoácidos presentes', fontsize=11)
ax.set_ylabel('Diversidad de Shannon (H)', fontsize=11)
ax.set_title('Cómo se reparten esos tipos', fontsize=13, fontweight='bold', pad=10)
ax.legend(fontsize=9, loc='lower right', framealpha=0.9)
fig.suptitle('Más tipos no significa más vida — importa el reparto',
fontsize=14, fontweight='bold', y=1.02)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.tight_layout()
plt.savefig('figuras/riqueza_vs_diversidad.png', dpi=200, bbox_inches='tight')
plt.show()
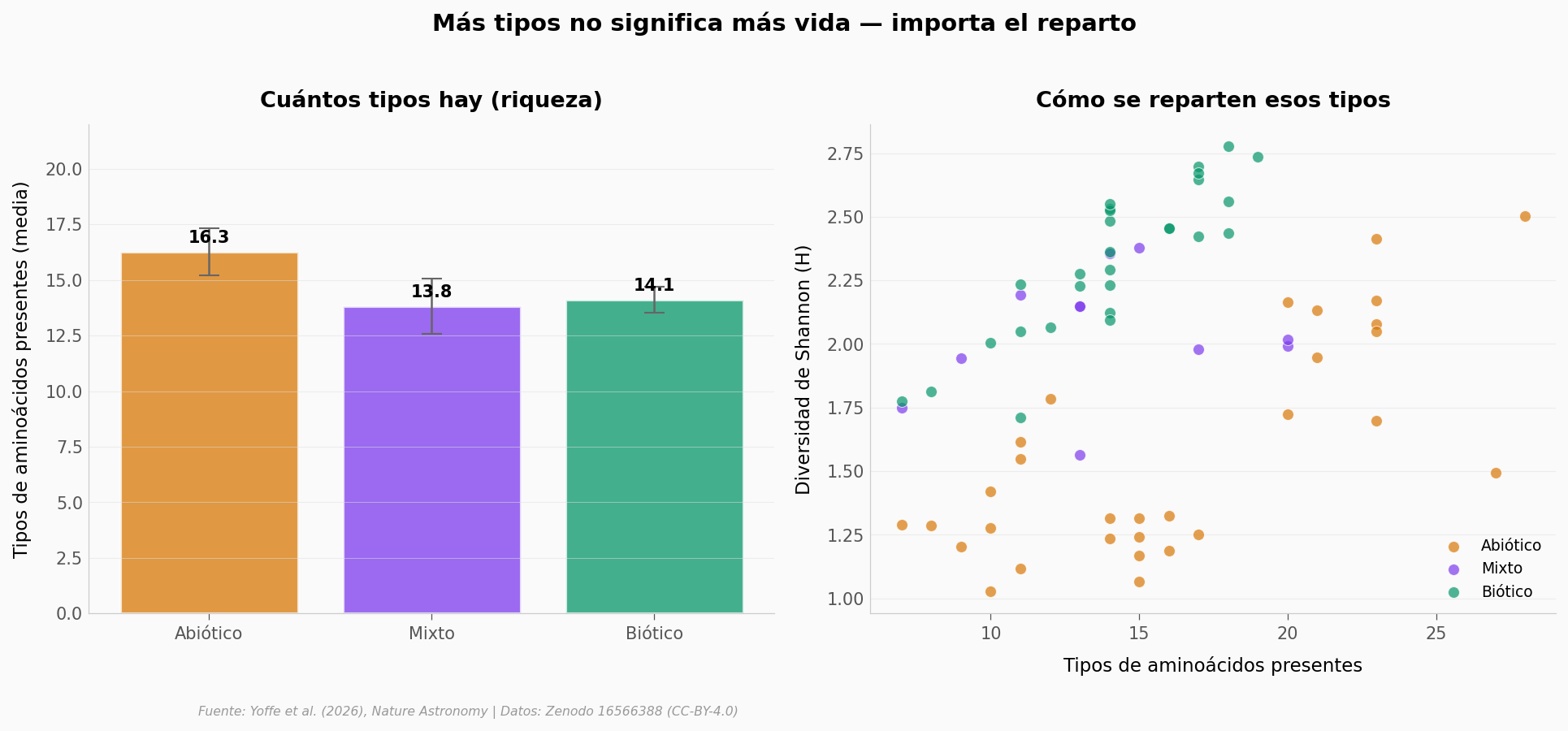
Lo que ven los datos: cuando los puntos ámbar tienen muchos tipos, su H se queda corto. Eso pasa porque dos aminoácidos — glicina y alanina — copan la fracción y dejan al resto en migajas. Cuando los puntos verdes tienen pocos tipos, su H ya es alta. La vida no acumula moléculas raras, las reparte.
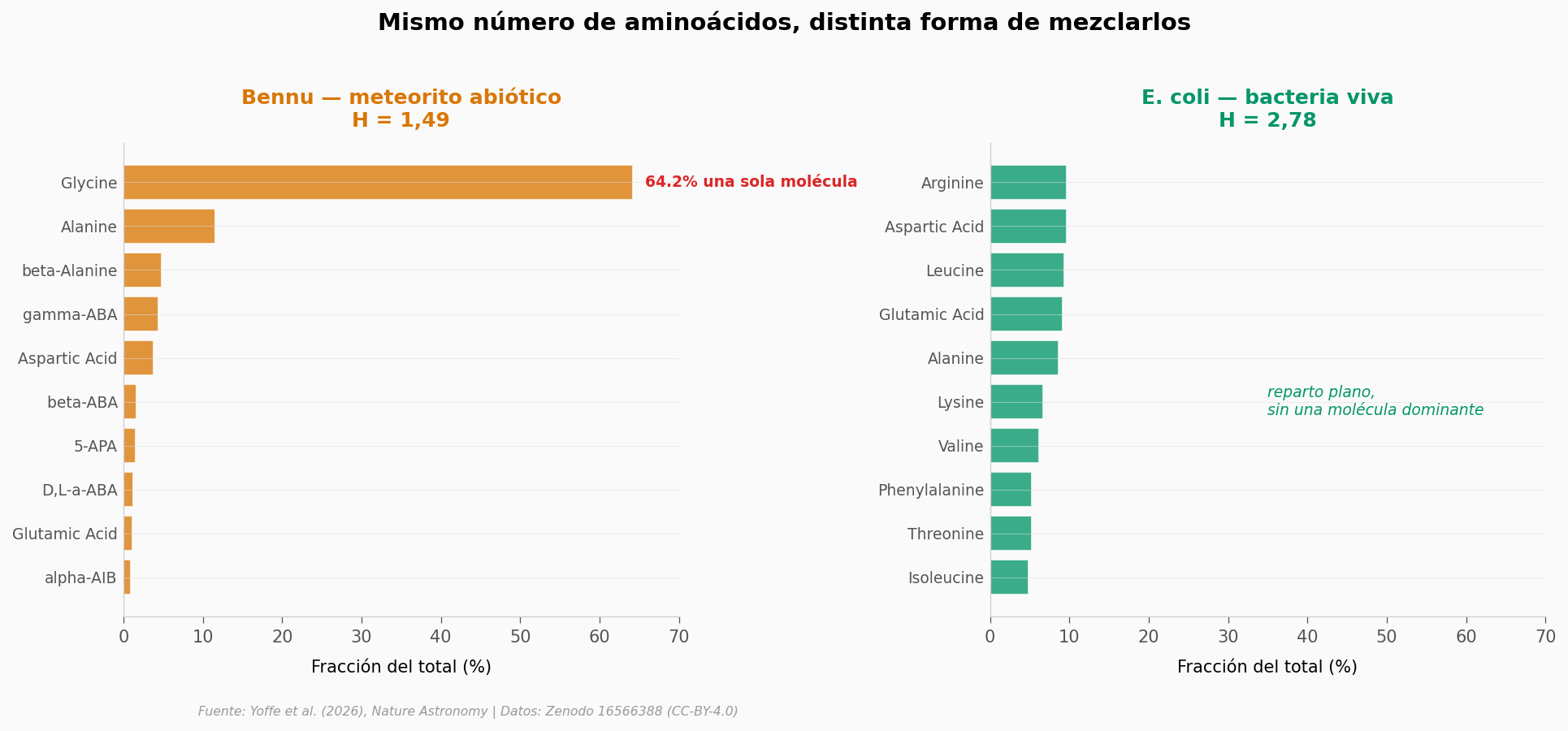
Para hacerlo concreto, miremos a Bennu — el cuerpo asteroidal del que la NASA trajo muestras en 2023, hoy guardado en Houston.
# Composición de Bennu vs la suma promedio de muestras bióticas
bennu_col = 'Bennu (CI) (Glavin+25)'
# Obtener abundancias: las dos primeras filas son meta (class label / stat model)
aa_data = aa_ab.iloc[2:].copy()
aa_data['Amino Acid'] = aa_data['Amino Acid'].astype(str)
# Bennu — tomar solo aminoácidos con valor numérico > 0
bennu = aa_data[['Amino Acid', bennu_col]].copy()
bennu[bennu_col] = pd.to_numeric(bennu[bennu_col], errors='coerce')
bennu = bennu.dropna()
bennu = bennu[bennu[bennu_col] > 0].copy()
bennu['frac'] = bennu[bennu_col] / bennu[bennu_col].sum()
bennu_top = bennu.sort_values('frac', ascending=False).head(10)
# Una muestra biótica representativa: E. Coli (paradigma de microbio vivo)
ecoli_col = [c for c in aa_ab.columns if 'E. Coli' in c or 'Coli' in c][0]
ecoli = aa_data[['Amino Acid', ecoli_col]].copy()
ecoli[ecoli_col] = pd.to_numeric(ecoli[ecoli_col], errors='coerce')
ecoli = ecoli.dropna()
ecoli = ecoli[ecoli[ecoli_col] > 0].copy()
ecoli['frac'] = ecoli[ecoli_col] / ecoli[ecoli_col].sum()
ecoli_top = ecoli.sort_values('frac', ascending=False).head(10)
fig, axes = plt.subplots(1, 2, figsize=(13, 5.5))
# Bennu
ax = axes[0]
y = np.arange(len(bennu_top))
ax.barh(y, bennu_top['frac'].values * 100,
color=COLOR_ABIOTIC, alpha=0.78, edgecolor='white', linewidth=1)
ax.set_yticks(y)
ax.set_yticklabels(bennu_top['Amino Acid'].values, fontsize=9)
ax.invert_yaxis()
ax.set_xlabel('Fracción del total (%)', fontsize=10)
ax.set_title(f'Bennu — meteorito abiótico\nH = 1,49', fontsize=12,
fontweight='bold', color=COLOR_ABIOTIC, pad=10)
ax.set_xlim(0, 70)
# Anotar dominancia de la glicina
gly_pct = bennu_top[bennu_top['Amino Acid'].str.contains('Gly', case=False)]['frac'].values
if len(gly_pct) > 0:
ax.text(gly_pct[0]*100 + 1.5, 0, f'{gly_pct[0]*100:.1f}% una sola molécula',
fontsize=9, color=COLOR_ALERTA, fontweight='bold', va='center')
# E. Coli
ax = axes[1]
y = np.arange(len(ecoli_top))
ax.barh(y, ecoli_top['frac'].values * 100,
color=COLOR_BIOTIC, alpha=0.78, edgecolor='white', linewidth=1)
ax.set_yticks(y)
ax.set_yticklabels(ecoli_top['Amino Acid'].values, fontsize=9)
ax.invert_yaxis()
ax.set_xlabel('Fracción del total (%)', fontsize=10)
ax.set_title('E. coli — bacteria viva\nH = 2,78', fontsize=12,
fontweight='bold', color=COLOR_BIOTIC, pad=10)
ax.set_xlim(0, 70)
ax.text(35, len(ecoli_top)/2, 'reparto plano,\nsin una molécula dominante',
fontsize=9, color=COLOR_BIOTIC, style='italic', va='center')
fig.suptitle('Mismo número de aminoácidos, distinta forma de mezclarlos',
fontsize=14, fontweight='bold', y=1.02)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.tight_layout()
plt.savefig('figuras/bennu_vs_ecoli.png', dpi=200, bbox_inches='tight')
plt.show()
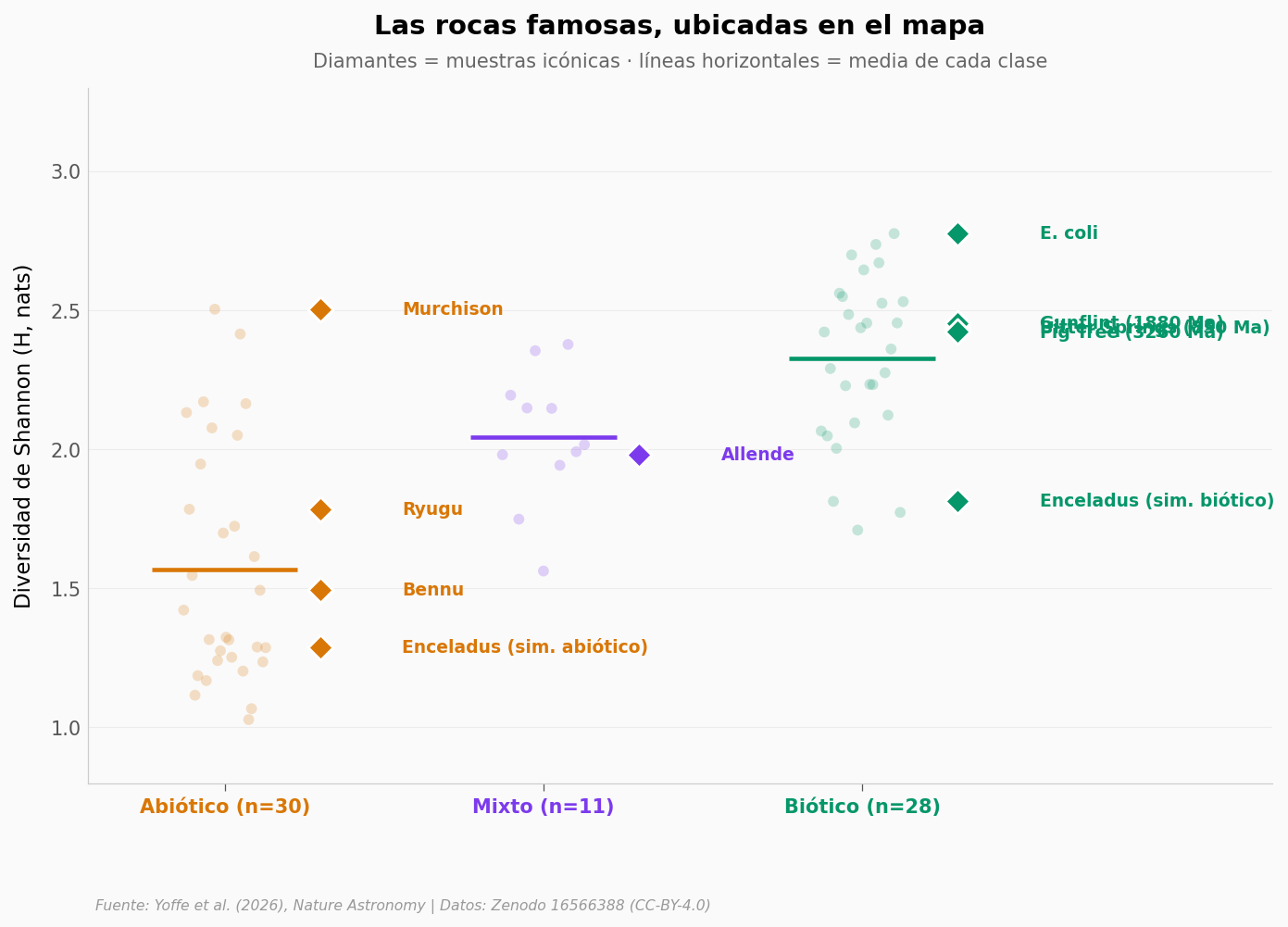
Y los iconos de la historia: ¿qué dicen?#
Miremos cómo caen las muestras que cualquier curso de astrobiología cita:
Bennu, Ryugu, Murchison: meteoritos carbonosos famosos.
Bitter Springs (~850 millones de años), Gunflint (~1880 Ma), Fig Tree (~3260 Ma): esteras microbianas fosilizadas — entre las pruebas más antiguas de vida en la Tierra.
Enceladus: una simulación de qué encontraríamos si la luna helada de Saturno tuviera vida vs si no.
# Marcar muestras icónicas sobre el strip plot principal
famous = {
'Bennu (CI) (Glavin+25)': ('abiotic', 'Bennu', 0.42),
'Ryugu (CI) (Glavin+22)': ('abiotic', 'Ryugu', 0.42),
'Murchison (CM2) (Glavin+11)': ('abiotic', 'Murchison', 0.42),
'Allende (CV3) (Cronin & Pizzarello)':('mixed', 'Allende', 1.82),
'Enceladus Abiotic': ('abiotic', 'Enceladus (sim. abiótico)', 0.42),
'Enceladus Biotic': ('biotic', 'Enceladus (sim. biótico)', 3.22),
'E. Coli (Bada+98 in Carroll+02)': ('biotic', 'E. coli', 3.22),
'Bitter Springs Chert (Brocks+03)': ('biotic', 'Bitter Springs (850 Ma)', 3.22),
'Gunflint Chert (Hahn+88)': ('biotic', 'Gunflint (1880 Ma)', 3.22),
'Fig Tree Chert (Sephton+04)': ('biotic', 'Fig Tree (3260 Ma)', 3.22),
}
# Mapear nombres del fact sheet a los del CSV (algunos varían)
samples_in_csv = aa['sample'].tolist()
match_map = {}
for key in famous:
# Búsqueda permisiva por substring
matches = [s for s in samples_in_csv if key.split(' (')[0] in s]
if matches:
match_map[matches[0]] = famous[key]
fig, ax = plt.subplots(figsize=(11, 6.5))
# Strip plot de fondo (puntos atenuados)
for c, label, color, pos in [('abiotic', 'Abiótico', COLOR_ABIOTIC, 0),
('mixed', 'Mixto', COLOR_MIXED, 1.4),
('biotic', 'Biótico', COLOR_BIOTIC, 2.8)]:
sub = aa[aa['class_label']==c]
n = len(sub)
np.random.seed(42)
x_jit = np.linspace(pos - 0.18, pos + 0.18, n)
np.random.shuffle(x_jit)
ax.scatter(x_jit, sub['H'].values, color=color, s=32, alpha=0.22,
edgecolors='none', zorder=3)
mean = sub['H'].mean()
ax.hlines(mean, pos - 0.32, pos + 0.32, color=color, lw=2.2, zorder=5)
# Anotar famous samples por encima
np.random.seed(7)
offset_x = 0
last_y_left, last_y_right = -10, -10
for csv_name, (cls, friendly, pos) in match_map.items():
row = aa[aa['sample']==csv_name].iloc[0]
H = row['H']
color = COLOR_ABIOTIC if cls=='abiotic' else (COLOR_BIOTIC if cls=='biotic' else COLOR_MIXED)
# Diamante grande
ax.scatter([pos], [H], marker='D', color=color, s=85,
edgecolors='white', linewidths=1.4, zorder=10)
# Texto a la derecha del punto
ax.annotate(friendly,
xy=(pos, H), xytext=(pos + 0.36, H),
fontsize=9, fontweight='bold', color=color,
va='center', ha='left', zorder=12)
ax.set_xticks([0, 1.4, 2.8])
ax.set_xticklabels(['Abiótico (n=30)', 'Mixto (n=11)', 'Biótico (n=28)'],
fontsize=10, fontweight='bold')
for tick, color in zip(ax.get_xticklabels(),
[COLOR_ABIOTIC, COLOR_MIXED, COLOR_BIOTIC]):
tick.set_color(color)
ax.set_ylabel('Diversidad de Shannon (H, nats)', fontsize=11)
ax.set_ylim(0.8, 3.3)
ax.set_xlim(-0.6, 4.6)
ax.set_title('Las rocas famosas, ubicadas en el mapa',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03,
'Diamantes = muestras icónicas · líneas horizontales = media de cada clase',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/famous_samples.png', dpi=200, bbox_inches='tight')
plt.show()
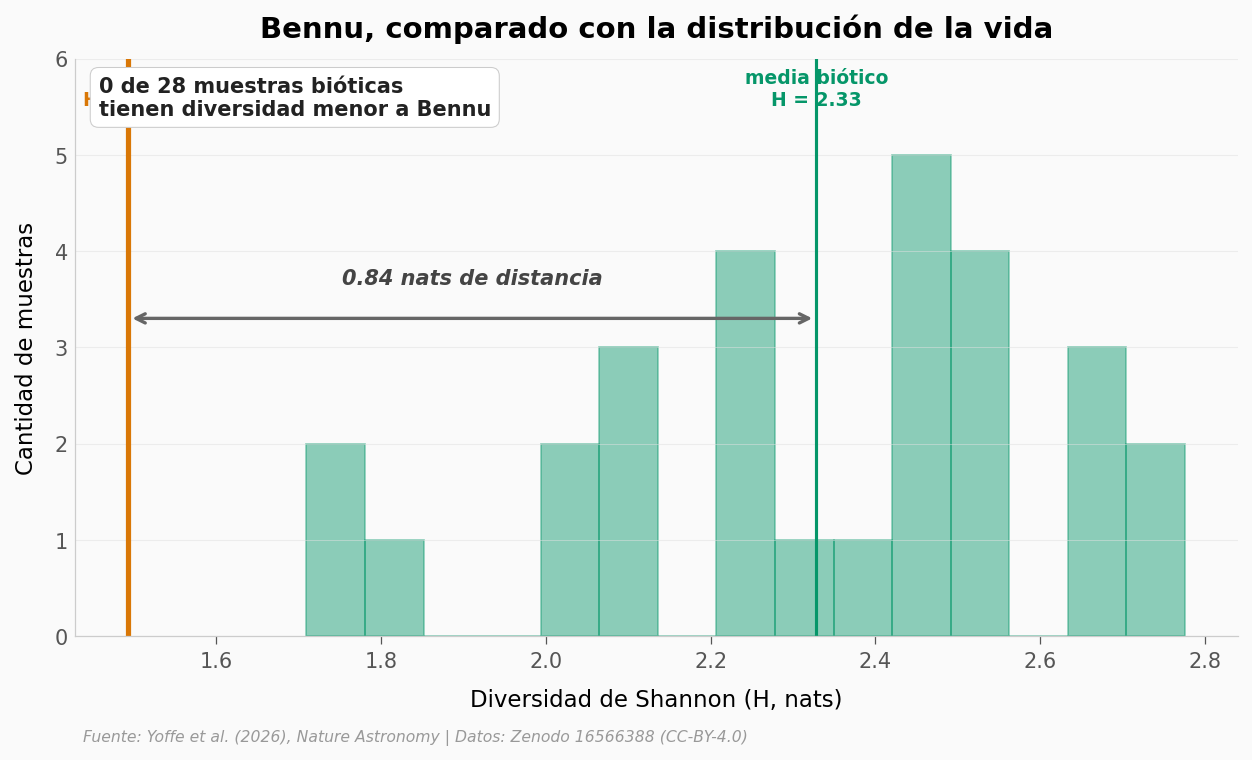
¿Qué tan rara sería una «vida» con la diversidad de Bennu?#
Pensemos al revés: si Bennu fuera vida, ¿cuántas muestras bióticas conocidas tendrían menos diversidad que él?
bennu_H = aa[aa['sample'].str.contains('Bennu')]['H'].values[0]
H_biotic = aa[aa['class_label']=='biotic']['H'].values
fig, ax = plt.subplots(figsize=(10, 5))
n, bins, _ = ax.hist(H_biotic, bins=15, color=COLOR_BIOTIC, alpha=0.45,
edgecolor=COLOR_BIOTIC, linewidth=0.8, label='Muestras bióticas (n=28)')
y_max = n.max() * 1.20
ax.set_ylim(0, y_max)
mean_biotic = H_biotic.mean()
ax.axvline(x=mean_biotic, color=COLOR_BIOTIC, linewidth=1.5, linestyle='-')
ax.text(mean_biotic, y_max*0.92, f'media biótico\nH = {mean_biotic:.2f}',
ha='center', fontsize=9, color=COLOR_BIOTIC, fontweight='bold')
ax.axvline(x=bennu_H, color=COLOR_ABIOTIC, linewidth=2.5)
ax.text(bennu_H, y_max*0.92, f'Bennu\nH = {bennu_H:.2f}',
ha='center', fontsize=9, color=COLOR_ABIOTIC, fontweight='bold')
# Flecha bidireccional
ax.annotate('', xy=(bennu_H, y_max*0.55), xytext=(mean_biotic, y_max*0.55),
arrowprops=dict(arrowstyle='<->', color='#666666', lw=1.6))
gap = mean_biotic - bennu_H
ax.text((bennu_H + mean_biotic)/2, y_max*0.61,
f'{gap:.2f} nats de distancia',
ha='center', fontsize=10, color='#444444', fontweight='bold',
style='italic')
# ¿Cuántos bióticos están por debajo de Bennu?
n_below = (H_biotic < bennu_H).sum()
ax.text(0.02, 0.97,
f'{n_below} de {len(H_biotic)} muestras bióticas\n'
f'tienen diversidad menor a Bennu',
transform=ax.transAxes, fontsize=10, color='#222222',
va='top', fontweight='bold',
bbox=dict(boxstyle='round,pad=0.4', facecolor='white', edgecolor='#CCCCCC'))
ax.set_xlabel('Diversidad de Shannon (H, nats)', fontsize=11)
ax.set_ylabel('Cantidad de muestras', fontsize=11)
ax.set_title('Bennu, comparado con la distribución de la vida',
fontsize=14, fontweight='bold', pad=10)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/anomalia_bennu.png', dpi=200, bbox_inches='tight')
plt.show()
Lo que los datos soportan#
Afirmación |
¿Soportada? |
Detalle |
|---|---|---|
Las muestras bióticas tienen mayor diversidad de Shannon que las abióticas (aminoácidos) |
✅ |
Diferencia de 0,76 nats (48,5%). Mann-Whitney p < 1e-7. Cohen’s d = 2,06 (efecto excepcionalmente grande). |
Las muestras abióticas tienen, en promedio, más tipos distintos de aminoácidos |
✅ |
16,3 vs 14,1. La diferencia es real, pero los abióticos están dominados por glicina/alanina. |
Bennu (asteroide) tiene baja diversidad pese a sus 27 tipos de aminoácidos |
✅ |
H = 1,49. Una sola molécula (glicina) ocupa el 64,2% del total. |
Las muestras «mixtas» caen en un punto intermedio |
✅ |
Media H = 2,04, entre abiótico (1,57) y biótico (2,33). |
El mismo patrón se replica en ácidos grasos |
⚠️ |
El paper lo reporta usando un marco probabilístico que ajusta cada muestra a una distribución y propaga incertidumbres. Con entropía de Shannon cruda sobre las abundancias publicadas, en este notebook no obtenemos el mismo resultado (p = 0,76 para ácidos grasos). Es una limitación honesta del enfoque simplificado — la firma diversa de la vida en ácidos grasos requiere la maquinaria completa del paper. |
La señal de diversidad refleja una firma biosintética fundamental |
— |
El paper lo plantea como inferencia; los datos sugieren la asociación, no prueban el mecanismo. |
Limitaciones que importan:
Estudio observacional. El diseño compara muestras pre-existentes; no es un experimento controlado. Los datos muestran asociación entre vida y reparto parejo, no causalidad mecanística.
Sesgo de muestreo. Las 28 muestras bióticas son mayoritariamente microbios cultivados y fósiles de la Tierra. Si la vida en otro lugar bioquímicamente exótica decide usar 2 o 3 aminoácidos dominantes, este test la marcaría como abiótica.
Réplica parcial. Confirmamos el resultado sólido para aminoácidos. Para ácidos grasos hace falta el modelo probabilístico del paper.
Ahora tú#
Tres preguntas para hurgar en los datos:
¿Murchison rompe la regla? Es un meteorito abiótico, pero su H = 2,50 está por encima de muchas muestras bióticas. ¿Qué aminoácidos lo dominan? Pista: filtra
aa_abpor la columna de Murchison y ordena las fracciones.¿Y si miramos la riqueza por separado? Repite el strip plot pero usando
n_presenten vez deH. ¿Las clases se siguen separando, o el solapamiento es total? Pista: copia la celda del strip plot y cambia'H'por'n_present'.¿Qué pasaría con la diversidad si Bennu tuviera 50% glicina en vez de 64%? Recalcula la entropía de Shannon redistribuyendo el exceso de glicina a los otros aminoácidos. Pista: ya tienes
bennucon las fracciones — modificafracy aplica-sum(p*log(p)).
# --- EXPERIMENTA AQUÍ ---
# Ejemplo: ¿qué pasa con la diversidad de Bennu si la glicina baja del 64% al 30%?
bennu_test = bennu[['Amino Acid', bennu_col]].copy().reset_index(drop=True)
bennu_test['frac_original'] = bennu_test[bennu_col] / bennu_test[bennu_col].sum()
# Redistribuir el exceso de glicina al resto, en proporción a sus fracciones actuales
target_gly = 0.30
gly_mask = bennu_test['Amino Acid'].str.contains('Gly', case=False, na=False)
gly_actual = bennu_test.loc[gly_mask, 'frac_original'].sum()
excess = gly_actual - target_gly
bennu_test['frac_nueva'] = bennu_test['frac_original'].copy()
bennu_test.loc[gly_mask, 'frac_nueva'] = target_gly
mask_resto = ~gly_mask
bennu_test.loc[mask_resto, 'frac_nueva'] = (
bennu_test.loc[mask_resto, 'frac_original']
+ excess * bennu_test.loc[mask_resto, 'frac_original'] / bennu_test.loc[mask_resto, 'frac_original'].sum()
)
# Shannon de ambas
def shannon(p):
p = p[p > 0]
return float(-(p * np.log(p)).sum())
H_original = shannon(bennu_test['frac_original'].values)
H_simulado = shannon(bennu_test['frac_nueva'].values)
print(f'Bennu original (glicina {gly_actual*100:.1f}%): H = {H_original:.3f}')
print(f'Bennu simulado (glicina {target_gly*100:.1f}%): H = {H_simulado:.3f}')
print(f'Diferencia: {H_simulado - H_original:+.3f} nats')
print(f'\n¿Cae todavía debajo de la media biótica ({H_biotic.mean():.3f})?',
'Sí' if H_simulado < H_biotic.mean() else 'No')
Bennu original (glicina 64.2%): H = 1.493
Bennu simulado (glicina 30.0%): H = 2.256
Diferencia: +0.762 nats
¿Cae todavía debajo de la media biótica (2.328)? Sí
Fuentes#
Paper: Molecular diversity as a biosignature
Nature Astronomy, 2026-05-11
Datos: Molecular diversity as a biosignature: Amino acids and fatty acid abundances and uncertainty .csv tables
Zenodo · CC-BY-4.0
16 afirmaciones del notebook verificadas contra estas fuentes
Créditos#
Paper: Yoffe et al. (2026), Nature Astronomy, DOI 10.1038/s41550-026-02864-z
Datos: Repositorio Zenodo 16566388, licencia CC-BY-4.0
Repositorio del Lab: github.com/Ciencia-a-Mordiscos/lab
Canal: Ciencia a Mordiscos