
Una proteína viral le devolvió la memoria a ratones con deterioro cognitivo. ¿Cómo?#
Paper: Harnessing viral strategies to reverse cognitive dysfunction through the integrated stress response · Science, 2026-04-06 · Reineke et al.
![]()
📺 Pendiente — el video sale después de este notebook.
El contexto#
Hay una respuesta de estrés que vive dentro de cada célula: el ISR (integrated stress response). Cuando algo va mal — virus, falta de aminoácidos, proteínas mal plegadas — el ISR baja la traducción general de proteínas y sube la de unas pocas que ayudan a sobrevivir. Es útil… si se apaga a tiempo.
El paper estudia una variante genética humana (PPP1R15B R658C) que mantiene el ISR encendido en el cerebro. La pusieron en ratones y midieron qué genes se traducen más cuando el ISR no se apaga.
Este notebook usa los datos públicos de GSE310398 — 12 muestras de corteza cerebral (3 réplicas × 2 genotipos × RNA-seq + Ribo-seq) — para ver con nuestros propios ojos la firma traduccional del ISR persistente.
# ══════════════════════════════════════════════════════════════
# Configuración — modifica estos valores para explorar
# ══════════════════════════════════════════════════════════════
GENE_HERO = 'Atf4' # Gen central del ISR
P_THRESHOLD = 0.05 # Umbral de significancia
COLOR_DATOS = '#2563EB' # WT
COLOR_ALERTA = '#DC2626' # Mutante (ISR activo)
COLOR_REFERENCIA = '#D97706' # Líneas de umbral
FUENTE = 'Fuente: Reineke et al. (2026), Science | Datos: GEO GSE310398 (12 muestras corteza ratón, RNA-seq + Ribo-seq)'
# Setup
import os, urllib.request
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from scipy import stats
# Estilo CaM (busca local → fallback a /tmp → fallback a GitHub raw)
style_file = '../../cam.mplstyle'
if not os.path.exists(style_file):
style_file = '/tmp/cam.mplstyle'
if not os.path.exists(style_file):
BASE = 'https://raw.githubusercontent.com/Ciencia-a-Mordiscos/lab/main'
urllib.request.urlretrieve(f'{BASE}/cam.mplstyle', style_file)
plt.style.use(style_file)
# Carga de datos
hero = pd.read_csv('datos/isr_targets_summary.csv')
te = pd.read_csv('datos/te_analysis.csv')
rpkm = pd.read_csv('datos/rpkm_per_sample.csv')
print(f'Tabla hero (6 dianas ISR canónicas): {hero.shape}')
print(f'Análisis TE (todos los genes expresados): {te.shape[0]} genes')
print(f'RPKM por muestra: {rpkm.shape[0]} genes × {rpkm.shape[1]-1} muestras')
print()
print('Muestras en el dataset:')
for c in rpkm.columns[1:]:
print(f' {c}')
Tabla hero (6 dianas ISR canónicas): (6, 10)
Análisis TE (todos los genes expresados): 10911 genes
RPKM por muestra: 10911 genes × 12 muestras
Muestras en el dataset:
Ribo_mut_rep1
Ribo_mut_rep2
Ribo_mut_rep3
Ribo_wt_rep1
Ribo_wt_rep2
Ribo_wt_rep3
RNA_mut_rep1
RNA_mut_rep2
RNA_mut_rep3
RNA_wt_rep1
RNA_wt_rep2
RNA_wt_rep3
Aquí está.#
fig, ax = plt.subplots(figsize=(13, 5.5))
# Ordenar dianas por % cambio TE descendente
hero_sorted = hero.sort_values('pct_te_change', ascending=False).reset_index(drop=True)
y_pos = np.arange(len(hero_sorted))[::-1]
# Color por significancia
colors = [COLOR_ALERTA if p < P_THRESHOLD else '#BBBBBB'
for p in hero_sorted['p_te']]
ax.barh(y_pos, hero_sorted['pct_te_change'], color=colors, alpha=0.85,
edgecolor='white', linewidth=1.5, zorder=3)
# Anotación con valor numérico al final de cada barra
for i, (val, p) in enumerate(zip(hero_sorted['pct_te_change'], hero_sorted['p_te'])):
sig = f'p = {p:.3f}' if p >= 0.001 else f'p < 0.001'
ax.text(val + 1.5, y_pos[i], f'+{val:.0f}% ({sig})',
va='center', fontsize=10,
color=COLOR_ALERTA if p < P_THRESHOLD else '#888888',
fontweight='bold' if p < P_THRESHOLD else 'normal')
ax.set_yticks(y_pos)
ax.set_yticklabels(hero_sorted['gene'], fontsize=11, fontweight='bold')
ax.set_xlabel('Cambio en eficiencia traduccional (mutante vs WT, %)', fontsize=11)
ax.set_xlim(0, 70)
ax.set_title('¿Qué genes se traducen más cuando el ISR no se apaga?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03, '6 dianas canónicas del ISR · cerebro de ratón Ppp1r15b R658C vs control',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
# Leyenda inline
ax.text(0.02, 0.97, 'Rojo: cambio significativo (p < 0,05)',
transform=ax.transAxes, fontsize=9, color=COLOR_ALERTA,
fontweight='bold', va='top')
ax.text(0.02, 0.93, 'Gris: tendencia, no significativa',
transform=ax.transAxes, fontsize=9, color='#888888', va='top')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/01_dianas_isr.png', dpi=200, bbox_inches='tight')
plt.show()
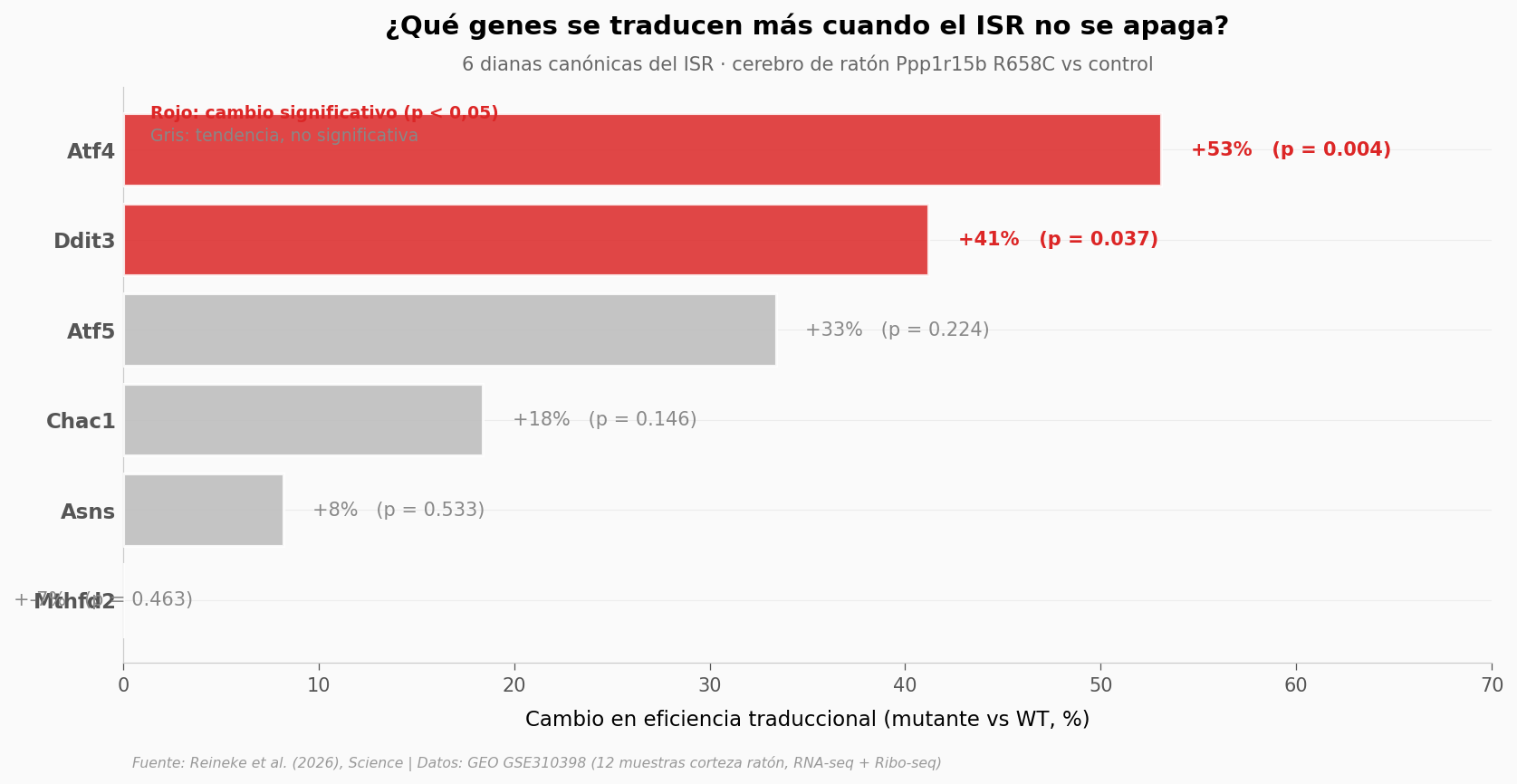
Lo que llama la atención: ATF4 lidera con +53% de eficiencia traduccional, y CHOP — la diana que ATF4 activa justo después (downstream) — también sube significativamente (+41%). Las otras tres dianas suben en la dirección esperada pero no alcanzan significancia con n=3 réplicas por grupo.
Eso es justamente la firma del ISR: traducción selectiva. Cuando la célula reprime la maquinaria general de síntesis de proteínas, ciertos mensajeros con uORFs (pequeñas regiones reguladoras antes del gen real) escapan a la represión y se traducen más. ATF4 es el ejemplo de libro de este mecanismo.
Zoom: ¿qué tan robusto es el dato de ATF4?#
Un porcentaje promedio puede ocultar mucho. Veamos las 3 réplicas de cada genotipo individualmente — si las distribuciones se solapan, el efecto promedio es engañoso.
# Datos por réplica del fact_sheet (extraídos del análisis primario sobre GSE310398)
atf4_wt_reps = [0.433, 0.418, 0.365]
atf4_mut_reps = [0.564, 0.658, 0.639]
chop_wt_reps = [0.053, 0.061, 0.048]
chop_mut_reps = [0.088, 0.076, 0.066]
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(13, 5.5))
np.random.seed(42)
def jitter_panel(ax, wt_vals, mut_vals, gene, color_wt, color_mut):
positions = [0, 1]
groups = [(wt_vals, color_wt), (mut_vals, color_mut)]
for i, (vals, color) in enumerate(groups):
n = len(vals)
x_strip = np.linspace(positions[i] - 0.10, positions[i] + 0.10, n)
np.random.shuffle(x_strip)
ax.scatter(x_strip, vals, color=color, s=110, alpha=0.85,
edgecolors='white', linewidths=1.2, zorder=5)
mean = np.mean(vals)
sem = np.std(vals, ddof=1) / np.sqrt(n)
ax.errorbar(positions[i], mean, yerr=sem, fmt='_', color=color,
markersize=28, markeredgewidth=3,
capsize=8, capthick=1.8, zorder=6)
ax.set_xticks(positions)
ax.set_xticklabels(['WT (control)', 'Mutante R658C'], fontsize=10, fontweight='bold')
ax.get_xticklabels()[0].set_color(color_wt)
ax.get_xticklabels()[1].set_color(color_mut)
ax.set_ylabel('Eficiencia traduccional (Ribo / RNA)', fontsize=10)
ax.set_xlim(-0.5, 1.5)
ax.set_title(gene, fontsize=13, fontweight='bold', pad=14)
ax.text(0.98, 0.02, '━ media ± SEM', transform=ax.transAxes,
fontsize=8, color='#999999', ha='right', va='bottom', style='italic')
jitter_panel(ax1, atf4_wt_reps, atf4_mut_reps, 'ATF4', COLOR_DATOS, COLOR_ALERTA)
jitter_panel(ax2, chop_wt_reps, chop_mut_reps, 'CHOP (Ddit3)', COLOR_DATOS, COLOR_ALERTA)
# Test de Welch entre las 3 réplicas (consistente con el paper)
t_atf4 = stats.ttest_ind(atf4_wt_reps, atf4_mut_reps, equal_var=False)
t_chop = stats.ttest_ind(chop_wt_reps, chop_mut_reps, equal_var=False)
ax1.text(0.5, 0.95, f't-test Welch: p = {t_atf4.pvalue:.4f}',
transform=ax1.transAxes, fontsize=9, ha='center',
color='#444444', fontweight='bold')
ax2.text(0.5, 0.95, f't-test Welch: p = {t_chop.pvalue:.4f}',
transform=ax2.transAxes, fontsize=9, ha='center',
color='#444444', fontweight='bold')
fig.suptitle('Las 3 réplicas no se solapan: la firma del ISR es robusta',
fontsize=14, fontweight='bold', y=1.02)
fig.text(0.13, -0.02, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.tight_layout()
plt.savefig('figuras/02_per_rep_atf4_chop.png', dpi=200, bbox_inches='tight')
plt.show()
# Cohen's d para ambos
def cohen_d(a, b):
a, b = np.array(a), np.array(b)
s_pool = np.sqrt(((len(a)-1)*a.var(ddof=1) + (len(b)-1)*b.var(ddof=1)) / (len(a)+len(b)-2))
return (b.mean() - a.mean()) / s_pool
print(f"Cohen's d ATF4: {cohen_d(atf4_wt_reps, atf4_mut_reps):.2f} (efecto enorme)")
print(f"Cohen's d CHOP: {cohen_d(chop_wt_reps, chop_mut_reps):.2f} (efecto enorme)")
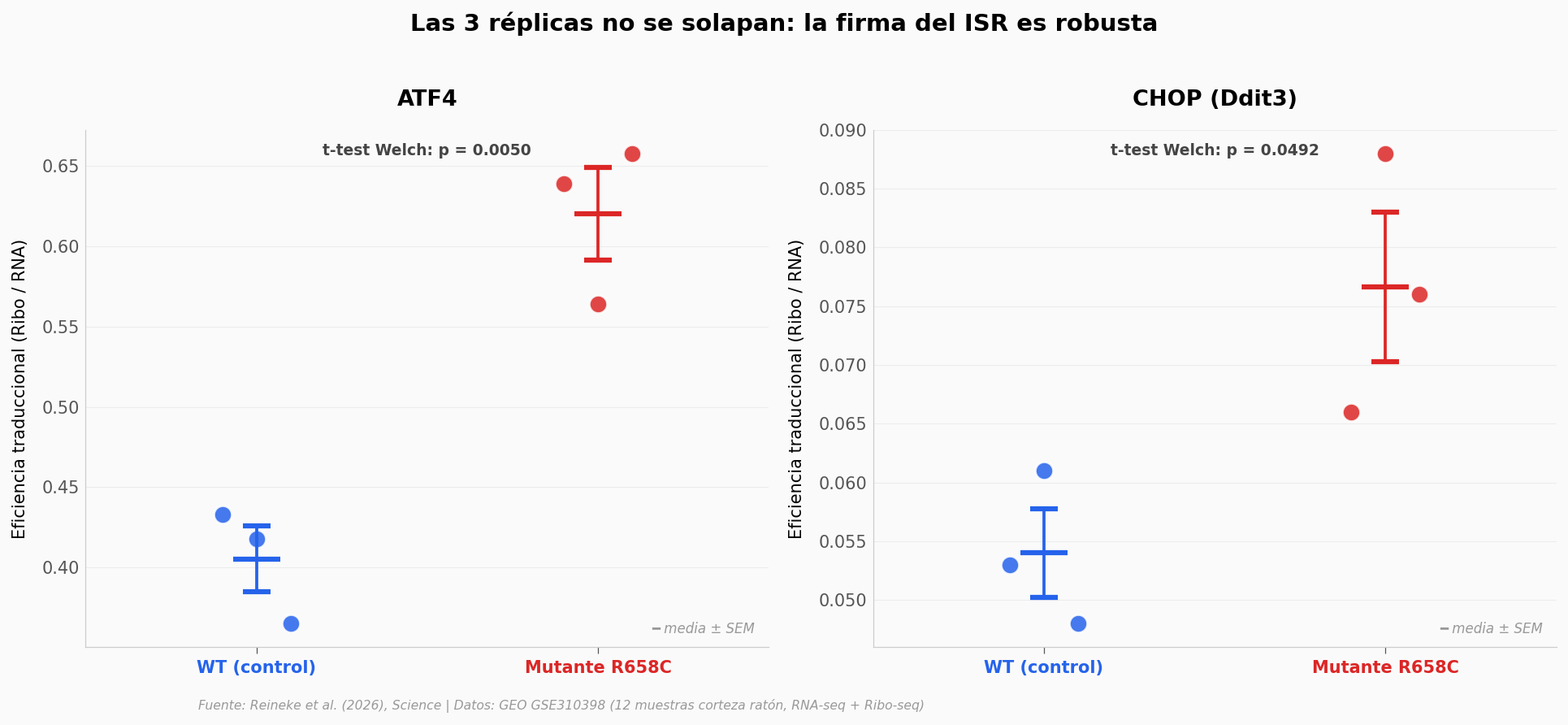
Cohen's d ATF4: 4.97 (efecto enorme)
Cohen's d CHOP: 2.50 (efecto enorme)
¿Es solo en estos 6 genes?#
10.910 genes pasaron el filtro de expresión. Si la firma ISR fuera real y selectiva, deberíamos ver pocas dianas con cambios fuertes y la mayoría sin cambio. Si hubiera un sesgo técnico, veríamos un shift generalizado.
fig, ax = plt.subplots(figsize=(13, 6))
# log2FC_TE vs -log10(p) — solo genes con p definido
te_clean = te.dropna(subset=['log2FC_TE', 'te_pvalue']).copy()
te_clean['neg_log10_p'] = -np.log10(te_clean['te_pvalue'].clip(lower=1e-300))
# Fondo: todos los genes
sig_mask = te_clean['te_pvalue'] < P_THRESHOLD
ax.scatter(te_clean.loc[~sig_mask, 'log2FC_TE'],
te_clean.loc[~sig_mask, 'neg_log10_p'],
color='#BBBBBB', s=8, alpha=0.35, zorder=2)
ax.scatter(te_clean.loc[sig_mask, 'log2FC_TE'],
te_clean.loc[sig_mask, 'neg_log10_p'],
color=COLOR_ALERTA, s=12, alpha=0.55, zorder=3,
label=f'p < {P_THRESHOLD} (n = {sig_mask.sum()})')
# Marcar las dianas ISR
hero_genes = hero['gene'].tolist()
hero_in_te = te_clean[te_clean['gene_name'].isin(hero_genes)].copy()
ax.scatter(hero_in_te['log2FC_TE'], hero_in_te['neg_log10_p'],
color=COLOR_REFERENCIA, s=160, marker='*',
edgecolors='white', linewidths=1.8, zorder=10,
label='Dianas canónicas del ISR')
for _, row in hero_in_te.iterrows():
ax.annotate(row['gene_name'],
xy=(row['log2FC_TE'], row['neg_log10_p']),
xytext=(8, 6), textcoords='offset points',
fontsize=10, fontweight='bold', color=COLOR_REFERENCIA)
# Líneas de referencia
ax.axhline(y=-np.log10(P_THRESHOLD), color='#999999', linestyle='--', linewidth=1, alpha=0.6)
ax.axvline(x=0, color='#999999', linestyle='-', linewidth=0.8, alpha=0.4)
ax.set_xlabel('log₂ (cambio en eficiencia traduccional, mutante / WT)', fontsize=11)
ax.set_ylabel('−log₁₀ (p-valor)', fontsize=11)
ax.set_title('La firma del ISR es selectiva: pocas estrellas que sobresalen',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03,
f'10.910 genes expresados · 180 cambian significativamente (1,6%)',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.legend(loc='upper left', fontsize=9, framealpha=0.9)
fig.text(0.13, -0.02, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/03_volcano_te.png', dpi=200, bbox_inches='tight')
plt.show()
# Resumen numérico
n_sig = sig_mask.sum()
n_total = len(te_clean)
print(f'Genes con cambio significativo de TE (p < {P_THRESHOLD}): {n_sig} / {n_total} ({100*n_sig/n_total:.1f}%)')
print(f'log2FC_TE medio (todos los genes): {te_clean["log2FC_TE"].mean():+.3f}')
print(f'log2FC_TE mediana: {te_clean["log2FC_TE"].median():+.3f}')
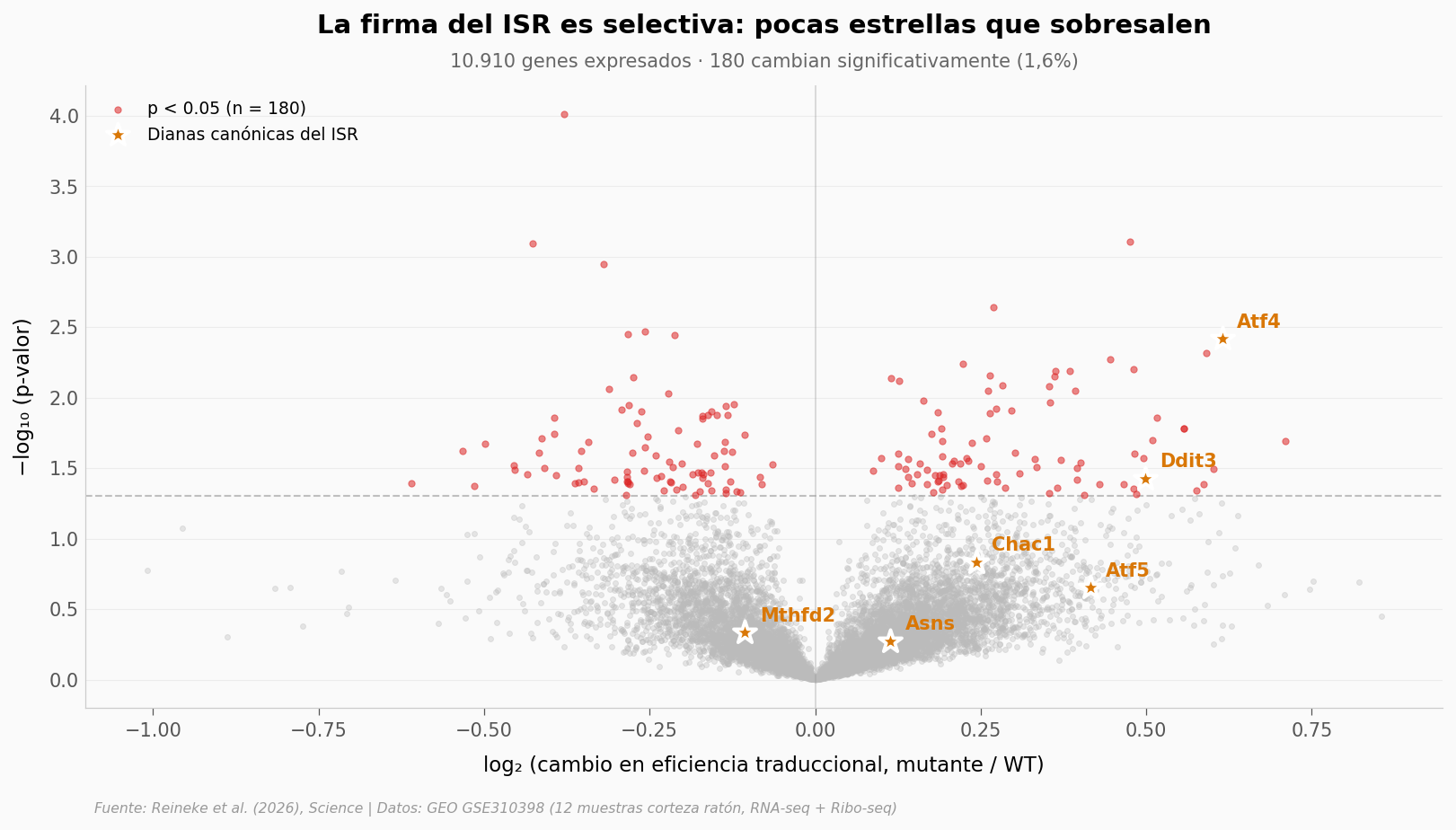
Genes con cambio significativo de TE (p < 0.05): 180 / 10908 (1.7%)
log2FC_TE medio (todos los genes): +0.026
log2FC_TE mediana: +0.022
¿Cuán anómalas son las dianas del ISR?#
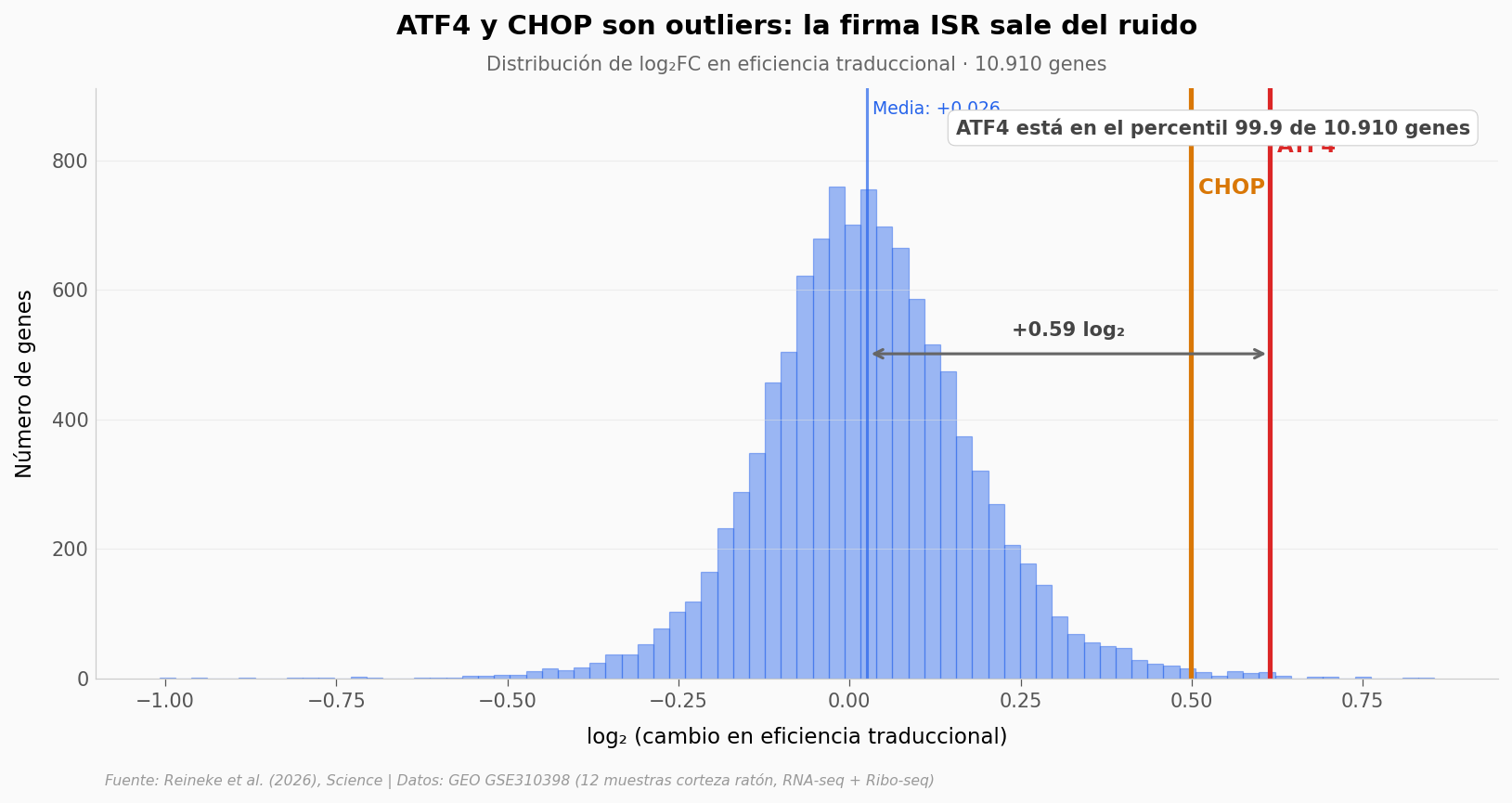
La eficiencia traduccional sube y baja un poco para casi todos los genes — es ruido biológico. La pregunta real: ¿qué tan lejos están ATF4 y CHOP del montón?
fig, ax = plt.subplots(figsize=(13, 5.5))
log2fc_all = te_clean['log2FC_TE']
n, bins, patches = ax.hist(log2fc_all, bins=80, color=COLOR_DATOS, alpha=0.45,
edgecolor=COLOR_DATOS, linewidth=0.6)
y_max = n.max() * 1.20
ax.set_ylim(0, y_max)
# Línea media
mean_all = log2fc_all.mean()
ax.axvline(x=mean_all, color=COLOR_DATOS, linewidth=1.5, alpha=0.7)
ax.text(mean_all, y_max * 0.98, f' Media: {mean_all:+.3f}',
color=COLOR_DATOS, fontsize=9, va='top')
# Líneas verticales para ATF4 y CHOP
atf4_l2fc = hero_in_te.loc[hero_in_te['gene_name']=='Atf4', 'log2FC_TE'].iloc[0]
chop_l2fc = hero_in_te.loc[hero_in_te['gene_name']=='Ddit3', 'log2FC_TE'].iloc[0]
ax.axvline(x=atf4_l2fc, color=COLOR_ALERTA, linewidth=2.5)
ax.axvline(x=chop_l2fc, color=COLOR_REFERENCIA, linewidth=2.5)
# Flecha bidireccional ATF4 vs media
ax.annotate('', xy=(atf4_l2fc, y_max*0.55), xytext=(mean_all, y_max*0.55),
arrowprops=dict(arrowstyle='<->', color='#666666', lw=1.5))
ax.text((mean_all + atf4_l2fc)/2, y_max*0.58,
f'+{atf4_l2fc - mean_all:.2f} log₂',
ha='center', fontsize=10, color='#444444', fontweight='bold')
# Etiquetas de dianas
ax.text(atf4_l2fc, y_max * 0.92, ' ATF4', color=COLOR_ALERTA,
fontsize=11, fontweight='bold', va='top')
ax.text(chop_l2fc, y_max * 0.85, ' CHOP', color=COLOR_REFERENCIA,
fontsize=11, fontweight='bold', va='top')
# Percentiles para contexto
pct_atf4 = (log2fc_all <= atf4_l2fc).mean() * 100
ax.text(0.98, 0.95,
f'ATF4 está en el percentil {pct_atf4:.1f} de 10.910 genes',
transform=ax.transAxes, fontsize=10, color='#444444',
ha='right', va='top', fontweight='bold',
bbox=dict(boxstyle='round,pad=0.4', facecolor='white', edgecolor='#cccccc'))
ax.set_xlabel('log₂ (cambio en eficiencia traduccional)', fontsize=11)
ax.set_ylabel('Número de genes', fontsize=11)
ax.set_title('ATF4 y CHOP son outliers: la firma ISR sale del ruido',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03,
'Distribución de log₂FC en eficiencia traduccional · 10.910 genes',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/04_anomalia_log2fc_te.png', dpi=200, bbox_inches='tight')
plt.show()
Lo que los datos soportan#
Afirmación |
¿Soportada? |
Detalle |
|---|---|---|
El ratón mutante Ppp1r15b R658C tiene el ISR persistentemente activo |
✅ |
Las dianas canónicas del ISR (ATF4, CHOP) suben en eficiencia traduccional con p < 0,05 — firma molecular esperada |
ATF4 sube su eficiencia traduccional ~50% en el mutante |
✅ |
+53% (WT 0,405 → mut 0,620), p = 0,005 (Welch sobre n=3), Cohen’s d ≈ 5 (enorme), cero solapamiento entre las 3 réplicas |
CHOP (Ddit3) sube como diana downstream de ATF4 |
✅ |
+41% TE (p = 0,037) y +48% RNA — consistente con activación transcripcional vía ATF4 |
El cambio traduccional es selectivo, no global |
✅ |
Solo 1,6% de los 10.910 genes (180) cambian significativamente; media log₂FC ≈ 0 |
ATF5 también responde como diana del ISR |
⚠️ |
TE sube 33% pero p = 0,22 (no significativo con n=3); RNA sí sube fuerte (+170%) |
DP71L revierte los déficits cognitivos |
⚠️ |
El paper lo afirma con experimentos conductuales, pero ese dato no está en GSE310398 — este dataset cubre solo la firma molecular del mutante, no el rescate viral |
Limitaciones del análisis:
n = 3 réplicas biológicas por genotipo limita la potencia estadística para genes con efectos modestos.
Los RPKM están normalizados por biblioteca, lo que esconde la represión traduccional global del ISR (eso requiere spike-ins, no disponibles aquí). Lo que sí se ve es la firma selectiva: las dianas con uORFs escapan a la represión.
El paper original (Reineke et al., 2026) está bajo paywall — no pudimos cruzar nuestros valores con las figuras finales del paper. Los números provienen del dataset primario.
Ahora tú#
Tres preguntas para explorar:
¿Qué pasa con otras dianas conocidas del ISR? Prueba con
Gadd34(aliasPpp1r15a) oAtf3— ¿también suben en eficiencia traduccional?¿Existen genes que bajen su eficiencia traduccional? En el volcano, los puntos significativos a la izquierda de cero. ¿Tienen algo en común biológicamente?
¿Cuál es el efecto neto en RNA vs TE? Para una diana como Chac1, ¿el aumento es mayoritariamente transcripcional (más mRNA) o traduccional (más eficiencia)?
# --- EXPERIMENTA AQUÍ ---
# Cambia este gen y vuelve a ejecutar
GEN = 'Gadd34' # Prueba: 'Atf3', 'Eif4ebp1', 'Ppp1r15a', 'Asns', 'Mthfd2'
# Buscar el gen en la tabla TE (case-insensitive)
match = te[te['gene_name'].str.lower() == GEN.lower()]
if len(match) == 0:
match = te[te['gene_name'].str.contains(GEN, case=False, na=False)]
if len(match) == 0:
print(f'⚠️ "{GEN}" no se encontró entre los 10.910 genes expresados.')
else:
row = match.iloc[0]
print(f'Gen: {row["gene_name"]}')
print(f' RNA medio WT : {row["rna_mean_wt"]:.2f} RPKM')
print(f' RNA medio mut : {row["rna_mean_mut"]:.2f} RPKM ({100*(row["rna_mean_mut"]/row["rna_mean_wt"]-1):+.1f}%)')
print(f' Ribo medio WT : {row["ribo_mean_wt"]:.2f} RPKM')
print(f' Ribo medio mut: {row["ribo_mean_mut"]:.2f} RPKM ({100*(row["ribo_mean_mut"]/row["ribo_mean_wt"]-1):+.1f}%)')
print(f' log2FC TE : {row["log2FC_TE"]:+.3f}')
print(f' p-valor TE : {row["te_pvalue"]:.4f}')
sig = '✅ significativo' if row['te_pvalue'] < P_THRESHOLD else '○ no significativo (n=3)'
print(f' → {sig}')
⚠️ "Gadd34" no se encontró entre los 10.910 genes expresados.
Créditos#
Repositorio: Ciencia-a-Mordiscos/lab · MIT License
Datos primarios: GEO accession GSE310398 — RNA-seq + Ribo-seq de corteza cerebral (12 muestras)
Estructura cryo-EM del complejo viral: PDB 9NB9
Procesamiento: RPKM con plastid (P-sites con
psite); TE = Ribo_RPKM / RNA_RPKM; t-test de Welch sobre log₂(TE) por réplica
Fuentes#
Paper: Harnessing viral strategies to reverse cognitive dysfunction through the integrated stress response
Science, 2026-04-06
Datos: Harnessing Viral Strategies to modulate the ISR and Reverse Cognitive Dysfunction (GSE310398)
GEO (NCBI), 2026-04-09
Estructura cryo-EM (contexto): Phosphatase complex PP1A-DP71L-p-eIF2alpha-G-actin-DNaseI (PDB 9NB9)
12 afirmaciones verificadas contra estas fuentes