
Dos pronúcleos compiten por el citoplasma en el zigoto#
54.7% vs 26.6%. Dos zigotos arrancan la vida con la misma información genética. Uno llega a nacer la mitad de las veces que el otro. La diferencia está en algo que la clínica de fecundación in vitro a veces ignora: que el embrión tenía un solo núcleo en lugar de dos.
Paper: Cytoplasmic competition between separate parental pronuclei in zygotes Nature, 2026 · Experimentos en zigotos de ratón con manipulación micromecánica + imaging cuantitativo
![]()
Video corto: Pendiente
El experimento#
Cuando un óvulo y un espermatozoide se fusionan, los cromosomas maternos y paternos quedan en dos compartimentos separados dentro de la misma célula: dos pronúcleos (PN). Es la última vez que mamá y papá viven en burbujas distintas antes de mezclarse para siempre.
A veces — por error de fertilización, o por manipulación en una clínica de fecundación in vitro — todos los cromosomas terminan en un solo pronúcleo (1PN biparental). Genéticamente debería dar igual: la información está toda ahí. Y sin embargo, casi la mitad de estos embriones falla en llegar a término.
Un equipo japonés midió tres cosas en zigotos de ratón: cuánto crece cada pronúcleo, qué pasa con las marcas químicas que regulan los genes, y a cuántos embriones les fue bien. Aquí están sus datos.
# ══════════════════════════════════════════════════════════════
# Configuración — modifica estos valores para explorar
# ══════════════════════════════════════════════════════════════
COLOR_2PN = '#2563EB' # Azul CaM — zigotos normales (2 pronúcleos)
COLOR_1PN = '#DC2626' # Rojo — zigotos 1PN biparentales (problema)
COLOR_P1PN = '#D97706' # Amber — P1PN, rescate experimental
COLOR_REF = '#BBBBBB' # Gris — referencias
FUENTE = 'Fuente: Mihajlović et al. (2026), Nature | Datos: Source Data MOESM10 + MOESM14'
import os, urllib.request
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from scipy import stats
# Estilo CaM — local si existe, fallback a raw GitHub
style_file = '../../cam.mplstyle'
if not os.path.exists(style_file):
style_file = '/tmp/cam.mplstyle'
if not os.path.exists(style_file):
urllib.request.urlretrieve(
'https://raw.githubusercontent.com/Ciencia-a-Mordiscos/lab/main/cam.mplstyle',
style_file,
)
plt.style.use(style_file)
os.makedirs('figuras', exist_ok=True)
# Carga
df_vol = pd.read_csv('datos/fig1b_pronuclear_volumes.csv')
df_his = pd.read_csv('datos/fig1e_histone_marks.csv')
df_dev = pd.read_csv('datos/fig5b_development_rates.csv')
print(f'Volúmenes pronucleares: {len(df_vol)} mediciones')
print(f'Marcas de histonas: {len(df_his)} mediciones')
print(f'Tasas de desarrollo: {len(df_dev)} filas (lotes × etapa × tipo)')
print()
print('Tipos de zigoto:', df_vol['zygote_type'].unique().tolist())
Volúmenes pronucleares: 170 mediciones
Marcas de histonas: 190 mediciones
Tasas de desarrollo: 50 filas (lotes × etapa × tipo)
Tipos de zigoto: ['2PN', '1PN']
Cuánto crece cada pronúcleo#
Aquí está.
fig, ax = plt.subplots(figsize=(13, 5.5))
# Tres grupos: maternal (2PN), paternal (2PN), biparental (1PN)
groups = [
('PN maternal\n(zigoto 2PN)', df_vol[(df_vol.zygote_type=='2PN') & (df_vol.pronucleus=='maternal_PN')]['volume_um3'].values, COLOR_2PN),
('PN paternal\n(zigoto 2PN)', df_vol[(df_vol.zygote_type=='2PN') & (df_vol.pronucleus=='paternal_PN')]['volume_um3'].values, COLOR_2PN),
('PN único\n(zigoto 1PN)', df_vol[(df_vol.zygote_type=='1PN') & (df_vol.pronucleus=='biparental_PN')]['volume_um3'].values, COLOR_1PN),
]
positions = [0, 1, 2.5]
np.random.seed(42)
for i, (label, vals, color) in enumerate(groups):
n = len(vals)
x = np.linspace(positions[i] - 0.13, positions[i] + 0.13, n)
np.random.shuffle(x)
ax.scatter(x, vals, color=color, s=42, alpha=0.65,
edgecolors='white', linewidths=0.5, zorder=5)
mean = vals.mean()
sem = vals.std(ddof=1) / np.sqrt(n)
ax.errorbar(positions[i], mean, yerr=sem, fmt='_', color=color,
markersize=24, markeredgewidth=3,
capsize=7, capthick=1.5, zorder=6)
# Anotar la media en negrita
ax.annotate(f'{mean:,.0f}'.replace(',', '.'), xy=(positions[i] + 0.20, mean),
fontsize=11, fontweight='bold', color=color, va='center')
# Línea de referencia: suma maternal + paternal del 2PN
suma_2pn = groups[0][1].mean() + groups[1][1].mean()
ax.axhline(y=suma_2pn, color=COLOR_REF, linestyle='--', linewidth=1.5, alpha=0.7, zorder=2)
ax.text(2.95, suma_2pn, f'suma m+p del 2PN ≈ {suma_2pn:,.0f} μm³'.replace(',', '.'),
fontsize=9, color='#888888', va='center')
ax.set_xticks(positions)
ax.set_xticklabels([g[0] for g in groups], fontsize=10, fontweight='bold')
for tick, color in zip(ax.get_xticklabels(), [COLOR_2PN, COLOR_2PN, COLOR_1PN]):
tick.set_color(color)
ax.set_ylabel('Volumen del pronúcleo (μm³)', fontsize=11)
ax.set_title('¿Cuánto crece cada pronúcleo en el zigoto?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03, 'Cuando hay un solo pronúcleo, ocupa lo que sumarían dos',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.text(0.98, 0.02, '━ media ± SEM', transform=ax.transAxes,
fontsize=8, color='#999999', ha='right', va='bottom', style='italic')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/volumenes_pronucleares.png', dpi=200, bbox_inches='tight')
plt.show()
# Reportar números clave
mPN = groups[0][1]; pPN = groups[1][1]; biPN = groups[2][1]
ratio = biPN.mean() / (mPN.mean() + pPN.mean())
print(f'PN maternal: media = {mPN.mean():.0f} μm³ (n={len(mPN)})')
print(f'PN paternal: media = {pPN.mean():.0f} μm³ (n={len(pPN)})')
print(f'PN biparental 1PN: media = {biPN.mean():.0f} μm³ (n={len(biPN)})')
print(f'Razón biPN / (m+p) = {ratio:.2f}x → el PN único ocupa el {100*ratio:.0f}% de lo que sumarían dos')
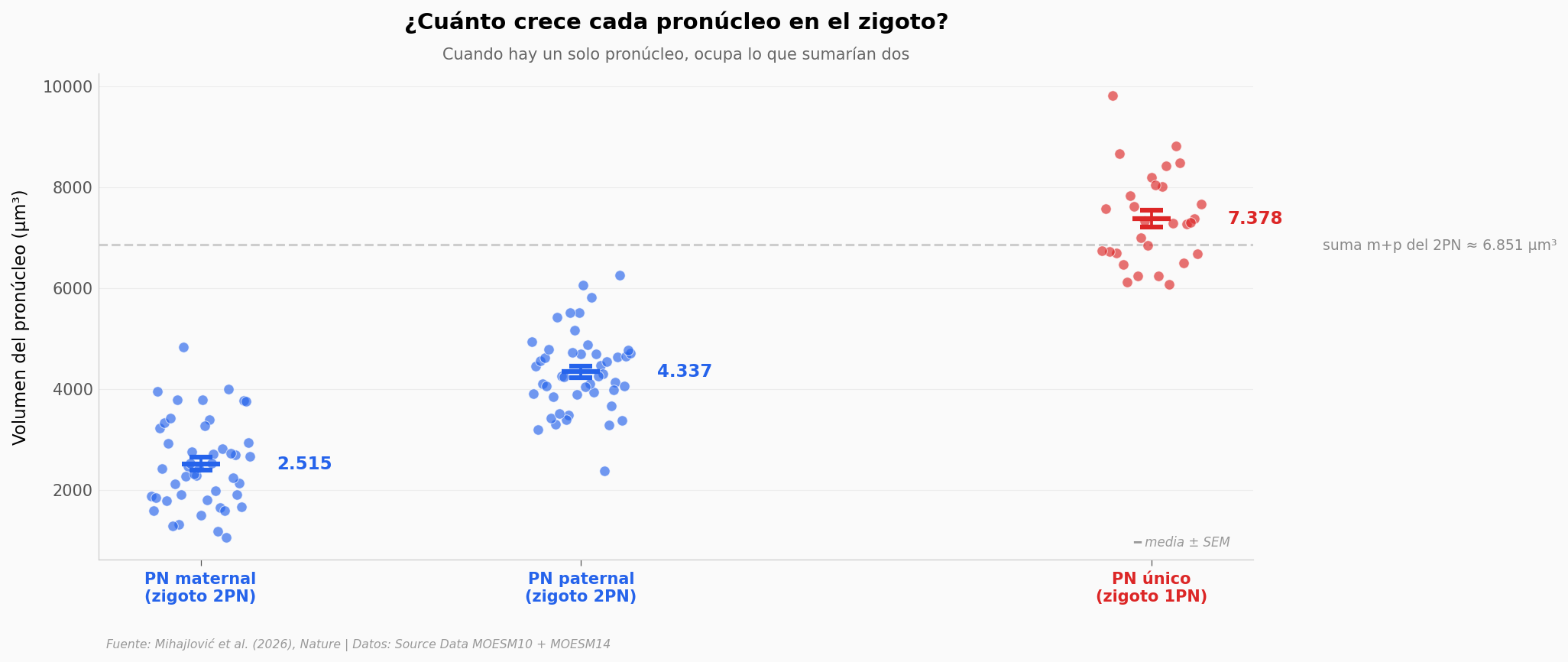
PN maternal: media = 2515 μm³ (n=47)
PN paternal: media = 4337 μm³ (n=47)
PN biparental 1PN: media = 7378 μm³ (n=29)
Razón biPN / (m+p) = 1.08x → el PN único ocupa el 108% de lo que sumarían dos
El pronúcleo paternal del zigoto normal es claramente más grande que el maternal — 4.337 frente a 2.515 μm³ en promedio (paternal 72% mayor, Cohen’s d = 1.84, n = 47 zigotos con ambos pronúcleos medidos). Es una asimetría conocida: el ADN del padre llega más descondensado. Pero el dato que importa: el pronúcleo único del 1PN ocupa el equivalente a sumar los dos del 2PN.
Esto es exactamente lo que predice la idea de «recursos citoplasmáticos finitos». Hay una cantidad limitada de algo (proteínas de importación nuclear, lípidos de membrana, lo que sea) y los pronúcleos compiten por ello. Con dos compartimentos compitiendo, cada uno se queda con su parte. Con uno solo, se lleva todo el lote.
¿Qué pasa con las marcas químicas del ADN?#
Que un pronúcleo crezca de más no es solo un problema de tamaño. Dentro del núcleo, las histonas — proteínas que empaquetan el ADN — llevan marcas químicas (metilaciones) que deciden qué genes se prenden y cuáles se apagan. Si el volumen cambia, las concentraciones de las enzimas que ponen y quitan esas marcas también cambian.
Veamos tres marcas distintas: H3K27me3, H3K4me3 y H3K9me3.
fig, ax = plt.subplots(figsize=(13, 5.5))
marks_order = ['H3K27me3', 'H3K4me3', 'H3K9me3']
positions_2pn = [0, 2, 4]
positions_1pn = [0.7, 2.7, 4.7]
np.random.seed(42)
for i, mark in enumerate(marks_order):
vals_2pn = df_his[(df_his['mark']==mark) & (df_his.zygote_type=='2PN')]['rel_intensity'].values
vals_1pn = df_his[(df_his['mark']==mark) & (df_his.zygote_type=='1PN')]['rel_intensity'].values
# 2PN scatter
x2 = np.linspace(positions_2pn[i] - 0.12, positions_2pn[i] + 0.12, len(vals_2pn))
np.random.shuffle(x2)
ax.scatter(x2, vals_2pn, color=COLOR_2PN, s=38, alpha=0.55,
edgecolors='white', linewidths=0.4, zorder=5)
ax.errorbar(positions_2pn[i], vals_2pn.mean(),
yerr=vals_2pn.std(ddof=1)/np.sqrt(len(vals_2pn)),
fmt='_', color=COLOR_2PN, markersize=22, markeredgewidth=3,
capsize=6, capthick=1.5, zorder=6)
# 1PN scatter
x1 = np.linspace(positions_1pn[i] - 0.12, positions_1pn[i] + 0.12, len(vals_1pn))
np.random.shuffle(x1)
ax.scatter(x1, vals_1pn, color=COLOR_1PN, s=38, alpha=0.55,
edgecolors='white', linewidths=0.4, zorder=5)
ax.errorbar(positions_1pn[i], vals_1pn.mean(),
yerr=vals_1pn.std(ddof=1)/np.sqrt(len(vals_1pn)),
fmt='_', color=COLOR_1PN, markersize=22, markeredgewidth=3,
capsize=6, capthick=1.5, zorder=6)
# Test Mann-Whitney
u, p = stats.mannwhitneyu(vals_2pn, vals_1pn, alternative='two-sided')
sig = '***' if p < 0.001 else ('**' if p < 0.01 else ('*' if p < 0.05 else 'n.s.'))
y_top = max(vals_2pn.max(), vals_1pn.max()) + 0.08
ax.plot([positions_2pn[i], positions_1pn[i]], [y_top, y_top], color='#666666', lw=0.8)
ax.text((positions_2pn[i] + positions_1pn[i])/2, y_top + 0.02,
sig, ha='center', fontsize=11, fontweight='bold', color='#666666')
ax.axhline(y=1.0, color=COLOR_REF, linestyle=':', linewidth=1, alpha=0.6, zorder=1)
# Inline labels
ax.text(-0.5, 1.7, 'Zigoto 2PN (normal)', fontsize=10, color=COLOR_2PN, fontweight='bold')
ax.text(-0.5, 1.55, 'Zigoto 1PN (un solo núcleo)', fontsize=10, color=COLOR_1PN, fontweight='bold')
ax.set_xticks([(p2+p1)/2 for p2, p1 in zip(positions_2pn, positions_1pn)])
ax.set_xticklabels(marks_order, fontsize=11, fontweight='bold')
ax.set_ylabel('Intensidad relativa (2PN media = 1.0)', fontsize=11)
ax.set_title('¿Qué marcas químicas se pierden en el zigoto 1PN?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03, 'Tres metilaciones de histonas, comparadas entre 2PN y 1PN',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.text(0.98, 0.02, '*** p<0.001 ** p<0.01 * p<0.05 n.s. no significativo (Mann-Whitney)',
transform=ax.transAxes, fontsize=8, color='#999999', ha='right', va='bottom', style='italic')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/marcas_histonas.png', dpi=200, bbox_inches='tight')
plt.show()
# Cohen's d
for mark in marks_order:
v2 = df_his[(df_his['mark']==mark) & (df_his.zygote_type=='2PN')]['rel_intensity'].values
v1 = df_his[(df_his['mark']==mark) & (df_his.zygote_type=='1PN')]['rel_intensity'].values
pooled = np.sqrt(((len(v2)-1)*v2.var(ddof=1) + (len(v1)-1)*v1.var(ddof=1)) / (len(v2)+len(v1)-2))
d = (v2.mean() - v1.mean()) / pooled
diff = (v1.mean() - v2.mean()) / v2.mean() * 100
print(f'{mark}: 2PN={v2.mean():.2f} 1PN={v1.mean():.2f} cambio={diff:+.1f}% Cohen\'s d={d:.2f}')
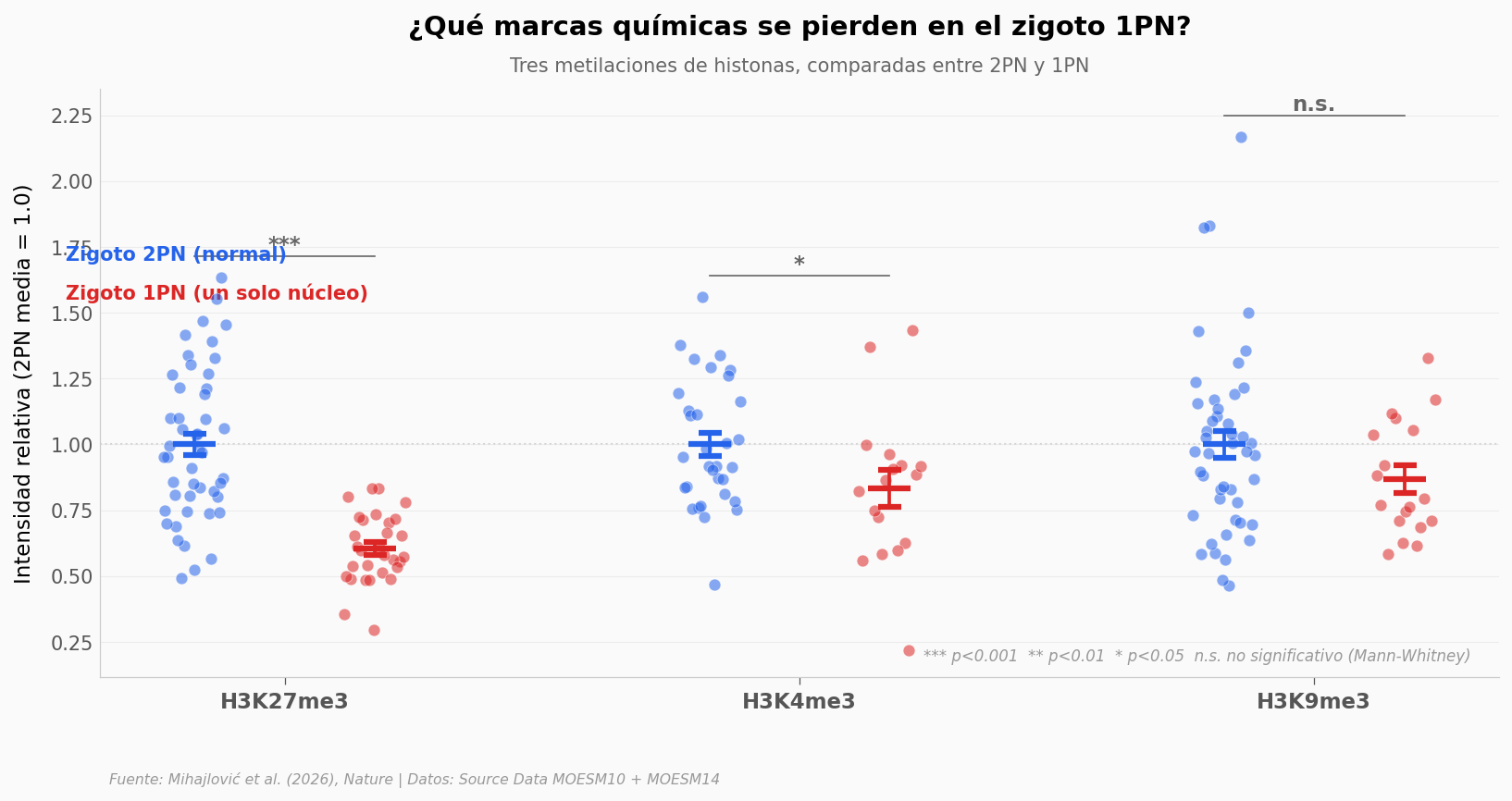
H3K27me3: 2PN=1.00 1PN=0.60 cambio=-39.6% Cohen's d=1.66
H3K4me3: 2PN=1.00 1PN=0.83 cambio=-16.8% Cohen's d=0.65
H3K9me3: 2PN=1.00 1PN=0.87 cambio=-13.2% Cohen's d=0.41
La marca que se cae a pedazos es H3K27me3 — el efecto es enorme (Cohen’s d = 1.66, prácticamente sin solapamiento entre grupos). Las otras dos se mueven menos: H3K4me3 baja un poco, H3K9me3 no se distingue del ruido.
H3K27me3 no es cualquier marca: es la responsable de mantener silenciados los genes que el embrión no debe encender todavía. Si se pierde, esos genes podrían activarse antes de tiempo. La pregunta entonces es directa: ¿cuántos de estos embriones sobreviven?
# Agregar lotes — usar promedio ponderado por n (regla 34)
def pooled_rate(df, stage, ztype):
sub = df[(df.developmental_stage==stage) & (df.zygote_type==ztype)]
return sub['successful'].sum() / sub['total'].sum(), sub['successful'].sum(), sub['total'].sum()
stages = ['2cell', 'full_term']
ztypes = [('2PN', COLOR_2PN, 'Zigoto 2PN\n(normal)'),
('1PN', COLOR_1PN, 'Zigoto 1PN\n(biparental)'),
('P1PN', COLOR_P1PN, 'P1PN\n(rescate)')]
fig, axes = plt.subplots(1, 2, figsize=(13, 5.5), sharey=True)
for ax_idx, stage in enumerate(stages):
ax = axes[ax_idx]
for i, (zt, color, label) in enumerate(ztypes):
rate, k, n = pooled_rate(df_dev, stage, zt)
# Wilson CI 95%
from math import sqrt
z = 1.96
denom = 1 + z**2/n
center = (rate + z**2/(2*n)) / denom
margin = z * sqrt(rate*(1-rate)/n + z**2/(4*n**2)) / denom
ci_low = max(0, center - margin)
ci_high = min(1, center + margin)
ax.bar(i, rate, color=color, alpha=0.85, width=0.6,
edgecolor='white', linewidth=1.5, zorder=3)
ax.errorbar(i, rate, yerr=[[rate-ci_low], [ci_high-rate]],
fmt='none', color='#333333', capsize=6, capthick=1.2, zorder=4)
ax.text(i, rate + 0.025, f'{100*rate:.1f}%', ha='center',
fontsize=11, fontweight='bold', color=color)
ax.text(i, -0.06, f'n={n}', ha='center', fontsize=8, color='#999999',
transform=ax.get_xaxis_transform())
ax.set_xticks(range(len(ztypes)))
ax.set_xticklabels([z[2] for z in ztypes], fontsize=9, fontweight='bold')
for tick, color in zip(ax.get_xticklabels(), [z[1] for z in ztypes]):
tick.set_color(color)
ax.set_ylim(0, 1.05)
if ax_idx == 0:
ax.set_ylabel('Tasa de éxito', fontsize=11)
title_es = 'Llegar a la división 2-células' if stage == '2cell' else 'Llegar a término'
ax.set_title(title_es, fontsize=13, fontweight='bold', pad=12)
fig.suptitle('¿Cuántos embriones lo logran?',
fontsize=14, fontweight='bold', y=1.02)
fig.text(0.5, 0.96, 'A las pocas horas casi todos sobreviven. Al final, la mitad de los 1PN se queda en el camino',
ha='center', fontsize=10, color='#666666')
fig.text(0.98, 0.04, 'Barras: IC 95% de Wilson sobre datos agrupados',
transform=fig.transFigure, fontsize=8, color='#999999', ha='right', style='italic')
fig.text(0.13, -0.02, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.tight_layout()
plt.savefig('figuras/tasas_desarrollo.png', dpi=200, bbox_inches='tight')
plt.show()
# Test χ² 2PN vs 1PN para full-term
ft_2pn = df_dev[(df_dev.developmental_stage=='full_term') & (df_dev.zygote_type=='2PN')]
ft_1pn = df_dev[(df_dev.developmental_stage=='full_term') & (df_dev.zygote_type=='1PN')]
table = np.array([[ft_2pn.successful.sum(), ft_2pn.total.sum() - ft_2pn.successful.sum()],
[ft_1pn.successful.sum(), ft_1pn.total.sum() - ft_1pn.successful.sum()]])
chi2, p, _, _ = stats.chi2_contingency(table)
print(f'2PN full-term: {ft_2pn.successful.sum()}/{ft_2pn.total.sum()} = {100*ft_2pn.successful.sum()/ft_2pn.total.sum():.1f}%')
print(f'1PN full-term: {ft_1pn.successful.sum()}/{ft_1pn.total.sum()} = {100*ft_1pn.successful.sum()/ft_1pn.total.sum():.1f}%')
print(f'χ² = {chi2:.2f}, p = {p:.2e}')
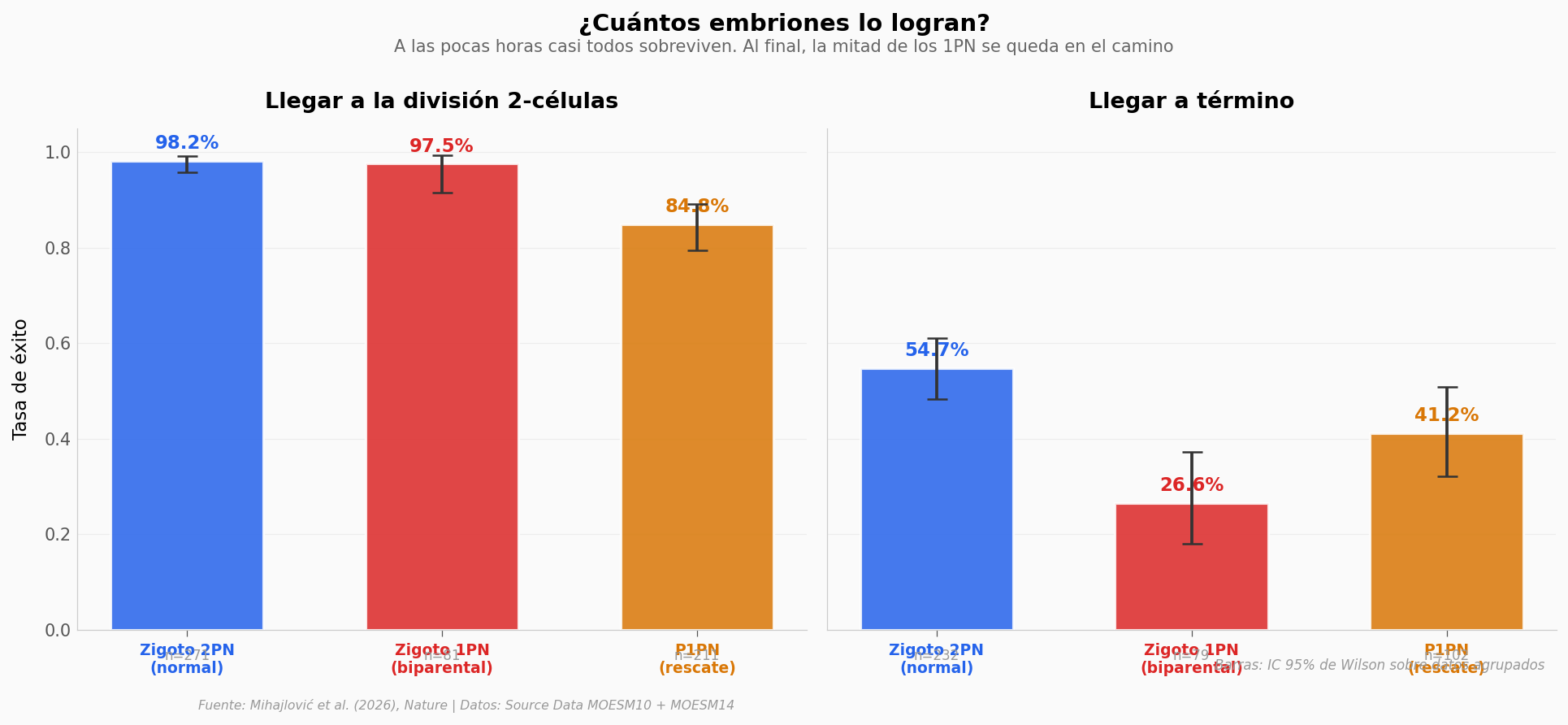
2PN full-term: 127/232 = 54.7%
1PN full-term: 21/79 = 26.6%
χ² = 17.62, p = 2.69e-05
¿Cuán raro es el pronúcleo 1PN comparado con los 2PN?#
A las pocas horas (etapa 2-células) ambos grupos van casi igual: 98% lo logra. Es al final del embarazo cuando se abre la brecha: 27% vs 55%, prácticamente la mitad. El rescate experimental P1PN — donde inyectan otro pronúcleo de un huevo no fecundado para forzar la competencia — recupera al 41%, partiendo la diferencia.
Veamos esa anomalía del volumen pronuclear de otra forma: ¿dónde cae el zigoto 1PN dentro de la distribución de zigotos 2PN normales?
fig, ax = plt.subplots(figsize=(11, 5))
vol_2pn_total = df_vol[(df_vol.zygote_type=='2PN') & (df_vol.pronucleus=='total_combined')]['volume_um3'].values
vol_1pn_biPN = df_vol[(df_vol.zygote_type=='1PN') & (df_vol.pronucleus=='biparental_PN')]['volume_um3'].values
n, bins, patches = ax.hist(vol_2pn_total, bins=22, color=COLOR_2PN, alpha=0.5,
edgecolor=COLOR_2PN, linewidth=0.8, label='Zigotos 2PN (suma m+p)')
y_max = n.max() * 1.25
ax.set_ylim(0, y_max)
# Media 2PN total
media_2pn = vol_2pn_total.mean()
ax.axvline(x=media_2pn, color=COLOR_2PN, linewidth=2, linestyle='--', alpha=0.85)
ax.text(media_2pn, y_max*0.92, f' media 2PN\n {media_2pn:,.0f} μm³'.replace(',', '.'),
fontsize=9, color=COLOR_2PN, fontweight='bold', va='top')
# Media 1PN biparental
media_1pn = vol_1pn_biPN.mean()
ax.axvline(x=media_1pn, color=COLOR_1PN, linewidth=2.5)
ax.text(media_1pn, y_max*0.92, f' media 1PN\n {media_1pn:,.0f} μm³'.replace(',', '.'),
fontsize=9, color=COLOR_1PN, fontweight='bold', va='top')
# Flecha
y_arrow = y_max * 0.55
ax.annotate('', xy=(media_1pn, y_arrow), xytext=(media_2pn, y_arrow),
arrowprops=dict(arrowstyle='<->', color='#666666', lw=1.5))
ax.text((media_2pn + media_1pn) / 2, y_arrow + y_max*0.03,
f'+{100*(media_1pn-media_2pn)/media_2pn:.0f}%',
ha='center', fontsize=12, fontweight='bold', color='#666666')
# Percentil del media-1PN dentro de la distribución 2PN
percentil = stats.percentileofscore(vol_2pn_total, media_1pn)
ax.text(0.98, 0.97,
f'El PN único del zigoto 1PN ({media_1pn:,.0f} μm³)\n cae en el percentil {percentil:.0f} de los 2PN totales'.replace(',', '.'),
transform=ax.transAxes, fontsize=10, color='#333333', ha='right', va='top',
bbox=dict(boxstyle='round,pad=0.4', facecolor='white', edgecolor='#CCCCCC'))
ax.set_xlabel('Volumen total de pronúcleo(s) (μm³)', fontsize=11)
ax.set_ylabel('Número de zigotos', fontsize=11)
ax.set_title('¿Dónde cae el zigoto 1PN dentro de la distribución 2PN?',
fontsize=14, fontweight='bold', pad=20)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/anomalia_volumen.png', dpi=200, bbox_inches='tight')
plt.show()
print(f'Volumen total 2PN: media = {media_2pn:.0f}, mediana = {np.median(vol_2pn_total):.0f} μm³ (n={len(vol_2pn_total)})')
print(f'Volumen único 1PN: media = {media_1pn:.0f}, mediana = {np.median(vol_1pn_biPN):.0f} μm³ (n={len(vol_1pn_biPN)})')
print(f'El 1PN cae en el percentil {percentil:.0f} de la distribución 2PN total')
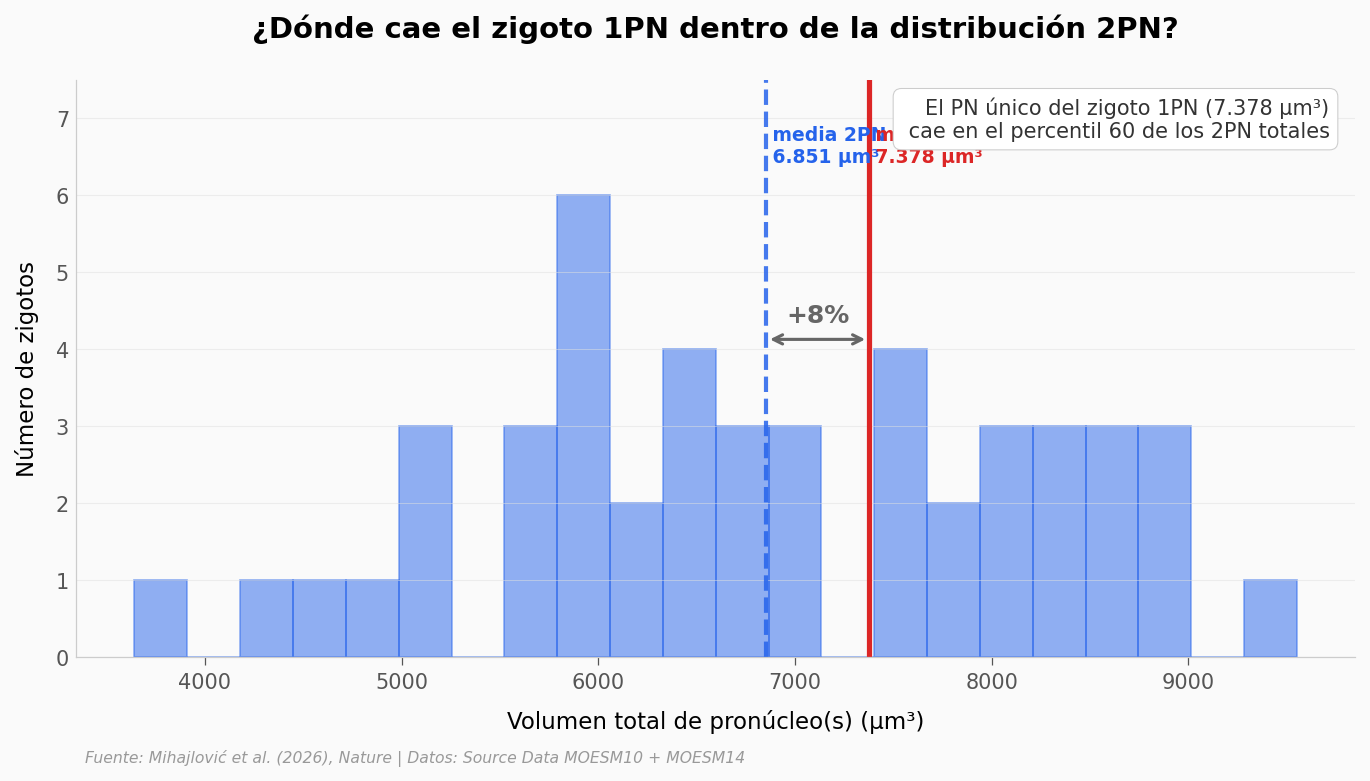
Volumen total 2PN: media = 6851, mediana = 6672 μm³ (n=47)
Volumen único 1PN: media = 7378, mediana = 7292 μm³ (n=29)
El 1PN cae en el percentil 60 de la distribución 2PN total
Lo que los datos soportan#
Afirmación |
¿Soportada? |
Detalle |
|---|---|---|
El PN paternal del 2PN es más grande que el maternal |
✅ |
4.337 vs 2.515 μm³ (paternal 72% mayor). Es asimetría conocida — el ADN paterno llega más descondensado. |
El PN único del 1PN ocupa lo que sumarían los dos del 2PN |
✅ |
7.378 vs 6.851 μm³ (razón 1.08x). Consistente con la hipótesis de recursos citoplasmáticos finitos. |
El zigoto 1PN pierde H3K27me3 |
✅ |
Cae ~40% (Cohen’s d = 1.66, p = 2×10⁻⁹, Mann-Whitney). Efecto muy grande. |
Las otras dos marcas también se pierden |
⚠️ |
H3K4me3 cae 17% (d = 0.65, p = 0.047) — efecto moderado, justo en el límite. H3K9me3 no se distingue del ruido (d = 0.41, p = 0.21). El paper agrupa como «marcas trimetiladas» pero el peso narrativo es H3K27me3. |
1PN tiene tasa de desarrollo a término reducida |
✅ |
27% vs 55% (χ² = 17.6, p = 3×10⁻⁵). Casi la mitad. |
El rescate P1PN recupera parcialmente |
✅ |
41% — intermedio entre 27% y 55%. Sugiere que la pérdida es reversible si se restaura la competencia. |
Hay un mecanismo de «competencia por citoplasma» entre pronúcleos |
⚠️ |
Los datos son consistentes con esa idea, pero el abstract usa suggesting y provides evidence of. Estos son experimentos en ratón con manipulación micromecánica: el siguiente paso sería identificar la molécula limitante concreta. |
Limitaciones:
Todos los experimentos son en zigotos de ratón — la traducción cuantitativa a humano requiere validación.
El rescate P1PN inyecta un pronúcleo de un huevo no fecundado: no es lo que ocurriría espontáneamente en una clínica.
El paper no identifica la molécula citoplasmática limitante — propone que existe, pero no la nombra.
El IC 95% de las tasas full-term es ancho para los grupos pequeños (1PN: n=79; P1PN: n=102 acumulado sobre 5 lotes); los lotes individuales son aún más ruidosos.
Ahora tú#
¿Y si miramos paternal vs maternal del 2PN como histogramas separados? ¿La asimetría se ve también en la dispersión, no solo en la media? Pista: filtra por
pronucleus == 'maternal_PN'ypaternal_PN, y usaax.hist()conalpha=0.5para superponer.¿Cómo varían las tasas full-term entre lotes experimentales del 1PN? Pista: agrupa
df_devporexperiment_batchparadevelopmental_stage == 'full_term'y revisa cuánto se mueve la tasa lote a lote.¿La pérdida de H3K27me3 correlaciona con el tamaño del pronúcleo único? Los datos del paper no permiten emparejar volúmenes con intensidades a nivel de embrión individual (vienen agregados), pero puedes comparar los promedios entre subgrupos para hacerte una idea.
# --- EXPERIMENTA AQUÍ ---
# Pregunta 2: ¿Cuánto varía la tasa full-term del 1PN entre los 5 lotes?
ft_1pn_lotes = df_dev[(df_dev.developmental_stage == 'full_term') & (df_dev.zygote_type == '1PN')].copy()
ft_1pn_lotes['rate_pct'] = 100 * ft_1pn_lotes['rate']
fig, ax = plt.subplots(figsize=(10, 4))
ax.bar(ft_1pn_lotes['experiment_batch'], ft_1pn_lotes['rate_pct'],
color=COLOR_1PN, alpha=0.7, edgecolor='white', linewidth=1.5)
for _, row in ft_1pn_lotes.iterrows():
ax.text(row['experiment_batch'], row['rate_pct'] + 1.5,
f"{row['rate_pct']:.0f}%\n(n={row['total']})",
ha='center', fontsize=9, color=COLOR_1PN)
# Línea de la tasa agregada
rate_agregada, k, n = pooled_rate(df_dev, 'full_term', '1PN')
ax.axhline(y=100*rate_agregada, color='#666666', linestyle='--', linewidth=1.2)
ax.text(ft_1pn_lotes['experiment_batch'].max() + 0.3, 100*rate_agregada,
f' agregada: {100*rate_agregada:.0f}%', fontsize=10, color='#666666', va='center')
ax.set_xlabel('Lote experimental')
ax.set_ylabel('Tasa full-term (%)')
ax.set_title('Tasa full-term del 1PN, lote por lote',
fontsize=13, fontweight='bold', pad=15)
ax.set_xticks(ft_1pn_lotes['experiment_batch'])
ax.set_ylim(0, max(ft_1pn_lotes['rate_pct'].max() * 1.2, 50))
plt.tight_layout()
plt.show()
print(f'\nDispersión entre lotes:')
print(f' rango: {ft_1pn_lotes.rate_pct.min():.1f}% a {ft_1pn_lotes.rate_pct.max():.1f}%')
print(f' media simple: {ft_1pn_lotes.rate_pct.mean():.1f}%')
print(f' media ponderada (correcta): {100*rate_agregada:.1f}%')
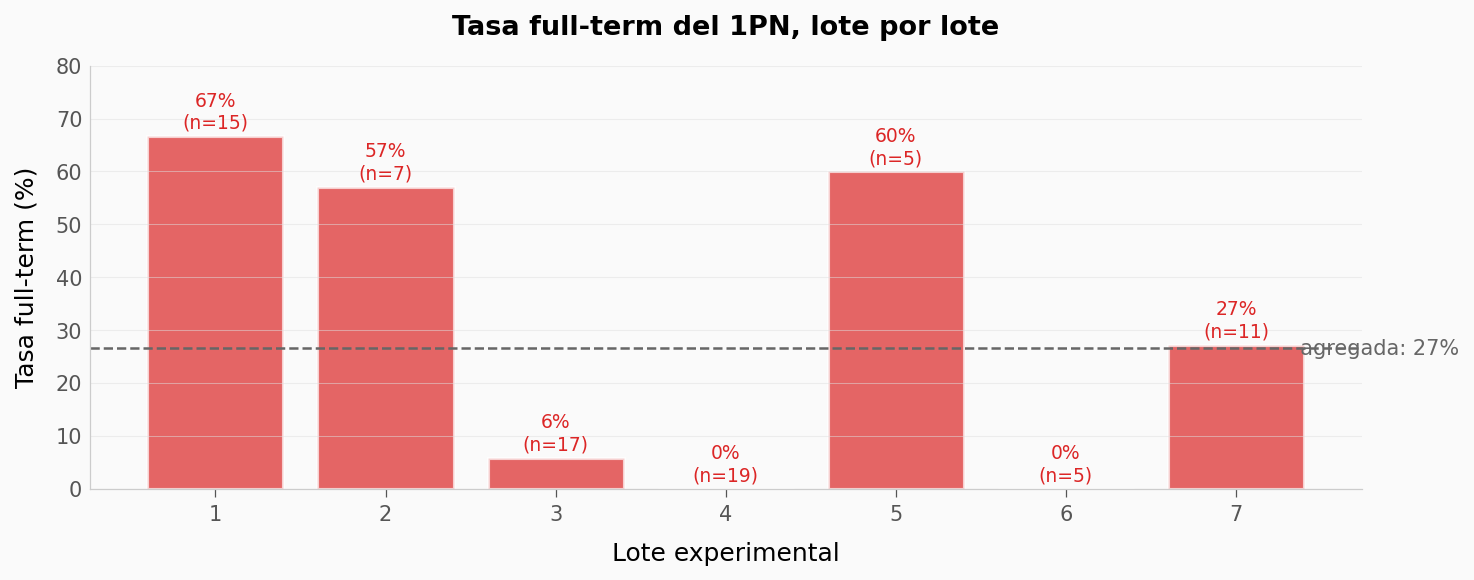
Dispersión entre lotes:
rango: 0.0% a 66.7%
media simple: 31.0%
media ponderada (correcta): 26.6%
Fuentes#
Paper: Cytoplasmic competition between separate parental pronuclei in zygotes
Nature, 2026-04-29
Source Data: Source Data Fig. 1 (volúmenes pronucleares + intensidades de histonas)
Source Data: Source Data Fig. 5 (tasas de desarrollo 2-cell y full-term)
16 afirmaciones del notebook verificadas contra estas fuentes
Generado por Ciencia a Mordiscos — Lab. Notebook reproducible: clona el repo, abre en Jupyter, ejecuta.