
128 genomas. Dos pistas inesperadas.#
Un equipo de 85 investigadores acaba de publicar el mayor conjunto de genomas indígenas americanos secuenciados hasta hoy: 128 individuos de 45 pueblos, repartidos entre México y la Patagonia.
La historia simple dice: los ancestros de los pueblos indígenas llegaron a América desde Siberia, se fueron extendiendo hacia el sur, y sus genomas reflejan esa expansión ordenada.
Los datos cuentan algo más complicado.
Paper: Castro e Silva et al. (2026), Nature Datos: Supplementary Tables 1–23 del paper (públicas, agregadas) Video: [Pendiente]
![]()
El estudio en una frase#
Secuenciaron 128 genomas completos (alta cobertura) de 45 poblaciones indígenas de 8 países latinoamericanos, los compararon con poblaciones antiguas y contemporáneas del resto del mundo, y buscaron pistas sobre cómo se pobló América.
Aquí vamos a mirar tres cosas con los datos agregados que publicaron:
¿Cuán marcadas son las huellas de cuellos de botella demográficos? — vía Runs of Homozygosity (ROH).
¿La distancia geográfica predice la distancia genética? — el modelo clásico de aislamiento por distancia.
¿Qué tan fuerte es la señal australasiática? — el rastro genético compartido con Papúa/Australia que nadie esperaba.
# ════════════════════════════════════════════════════════════════
# Configuración — modifica estos valores para explorar
# ════════════════════════════════════════════════════════════════
FUENTE = 'Fuente: Castro e Silva et al. (2026), Nature | Datos: Supplementary MOESM3'
COLOR_DATOS = '#2563EB' # azul CaM — indígenas americanos
COLOR_ALERTA = '#DC2626' # rojo — contrastes
COLOR_SECUNDARIO = '#059669' # verde — segundo grupo
COLOR_REFERENCIA = '#D97706' # ámbar — referencia / umbral
COLOR_CONTEXTO = '#BBBBBB' # gris — contexto
# ════════════════════════════════════════════════════════════════
# Imports
# ════════════════════════════════════════════════════════════════
import os, urllib.request
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from scipy import stats
# Estilo CaM (local primero, GitHub raw como fallback)
BASE = 'https://raw.githubusercontent.com/Ciencia-a-Mordiscos/lab/main'
style_file = '../../cam.mplstyle'
if not os.path.exists(style_file):
style_file = '/tmp/cam.mplstyle'
if not os.path.exists(style_file):
urllib.request.urlretrieve(f'{BASE}/cam.mplstyle', style_file)
plt.style.use(style_file)
# ════════════════════════════════════════════════════════════════
# Carga de datos
# ════════════════════════════════════════════════════════════════
genomas = pd.read_csv('datos/tabla1_128_genomas.csv')
roh = pd.read_csv('datos/tabla4_roh_mundial.csv')
dist = pd.read_csv('datos/tabla7_distancias.csv')
austral = pd.read_csv('datos/tabla12_australasian_counts.csv')
# Mapa 1000G population → superpoblación (codes estándar 1000 Genomes)
SUPERPOP_1000G = {
# AFR
'YRI':'AFR','LWK':'AFR','GWD':'AFR','MSL':'AFR','ESN':'AFR','ACB':'AFR','ASW':'AFR',
# EUR
'CEU':'EUR','TSI':'EUR','FIN':'EUR','GBR':'EUR','IBS':'EUR',
# EAS
'CHB':'EAS','JPT':'EAS','CHS':'EAS','CDX':'EAS','KHV':'EAS',
# SAS
'GIH':'SAS','PJL':'SAS','BEB':'SAS','STU':'SAS','ITU':'SAS',
# AMR admixed (hispano/latino del 1000G, NO indígenas)
'MXL':'AMR_admixed','PUR':'AMR_admixed','CLM':'AMR_admixed','PEL':'AMR_admixed',
}
roh['SuperPop'] = roh['Population'].map(SUPERPOP_1000G).fillna('Indigenous_American')
print(f'128 genomas nuevos: {len(genomas)} individuos de {genomas["Population"].nunique()} poblaciones')
print(f'Países: {", ".join(sorted(genomas["Country"].unique()))}')
print()
print('Tabla de ROH — 3.710 individuos mundiales:')
print(roh.groupby('SuperPop').agg(n=('FROH','size'), median=('FROH','median'), mean=('FROH','mean')).round(4))
print()
print(f'Distancias por pares: {len(dist)} comparaciones ({", ".join(dist["Comparison"].unique())})')
print(f'Señal australasiática: {austral["n_sig_ancient"].notna().sum()} pob. antiguas vs {len(austral)} contemporáneas')
128 genomas nuevos: 128 individuos de 45 poblaciones
Países: Argentina, Bolivia, Brazil, Colombia, Ecuador, Mexico, Paraguay, Peru
Tabla de ROH — 3.710 individuos mundiales:
n median mean
SuperPop
AFR 661 0.0152 0.0166
AMR_admixed 347 0.0373 0.0440
EAS 504 0.0516 0.0532
EUR 503 0.0436 0.0448
Indigenous_American 1206 0.0535 0.0688
SAS 489 0.0412 0.0486
Distancias por pares: 1378 comparaciones (SA-SA, NA-SA, NA-NA)
Señal australasiática: 36 pob. antiguas vs 114 contemporáneas
Cuellos de botella: ¿cuánto se nota?#
Cuando una población se contrae en el pasado (guerras, epidemias, migraciones pequeñas), deja una huella en el genoma. Los segmentos de ADN idénticos heredados de un ancestro común se vuelven largos y numerosos — se llaman Runs of Homozygosity (ROH).
El FROH es la fracción del genoma cubierta por ROH mayores a 1,5 Mb: cuanto más alto, más marcados son los cuellos de botella históricos.
Aquí está.
fig, ax = plt.subplots(figsize=(13, 5.5))
# Orden + colores por superpoblación
orden = ['AFR', 'EUR', 'EAS', 'SAS', 'AMR_admixed', 'Indigenous_American']
labels = ['Africa\n(AFR)', 'Europa\n(EUR)', 'Asia Este\n(EAS)', 'Asia Sur\n(SAS)', 'Mestiza AMR\n(1000G)', 'Indígena\namericana']
colores = [COLOR_CONTEXTO, COLOR_CONTEXTO, COLOR_CONTEXTO, COLOR_CONTEXTO, COLOR_SECUNDARIO, COLOR_DATOS]
np.random.seed(42)
positions = np.arange(len(orden))
for i, grupo in enumerate(orden):
vals = roh.loc[roh['SuperPop'] == grupo, 'FROH'].values
n = len(vals)
# Jitter reproducible
x_jitter = np.linspace(positions[i] - 0.18, positions[i] + 0.18, n)
np.random.shuffle(x_jitter)
ax.scatter(x_jitter, vals, color=colores[i], s=12, alpha=0.35,
edgecolors='white', linewidths=0.2, zorder=3)
mediana = np.median(vals)
q1, q3 = np.percentile(vals, [25, 75])
ax.errorbar(positions[i], mediana, yerr=[[mediana - q1], [q3 - mediana]],
fmt='_', color=colores[i], markersize=28, markeredgewidth=3.5,
capsize=8, capthick=2, zorder=6)
ax.set_xticks(positions)
ax.set_xticklabels(labels, fontsize=10)
for tick, color in zip(ax.get_xticklabels(), colores):
tick.set_color(color)
tick.set_fontweight('bold')
ax.set_ylabel('FROH (fracción del genoma en ROH > 1,5 Mb)', fontsize=11)
ax.set_title('¿Qué grupo carga más huellas de cuellos de botella?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03, 'FROH por individuo en 6 grupos mundiales (n = 3.710)',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.text(0.98, 0.02, '━ mediana, barras = rango intercuartílico (Q1–Q3)',
transform=ax.transAxes, fontsize=8, color='#999999',
ha='right', va='bottom', style='italic')
ax.set_ylim(-0.005, max(roh['FROH']) * 1.05)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.tight_layout()
plt.savefig('figuras/roh_hero.png', dpi=200, bbox_inches='tight')
plt.show()
# Comparación Indígenas vs Europeos
ind_froh = roh.loc[roh['SuperPop'] == 'Indigenous_American', 'FROH'].values
eur_froh = roh.loc[roh['SuperPop'] == 'EUR', 'FROH'].values
u_stat, p_mw = stats.mannwhitneyu(ind_froh, eur_froh, alternative='greater')
pooled_sd = np.sqrt(((len(ind_froh) - 1) * ind_froh.var(ddof=1) + (len(eur_froh) - 1) * eur_froh.var(ddof=1)) / (len(ind_froh) + len(eur_froh) - 2))
d = (ind_froh.mean() - eur_froh.mean()) / pooled_sd
print(f'Mediana FROH — Indígena americana: {np.median(ind_froh):.4f}')
print(f'Mediana FROH — Europea (EUR): {np.median(eur_froh):.4f}')
print(f'Razón de medianas: {np.median(ind_froh)/np.median(eur_froh):.2f}x')
print(f'Mann-Whitney U (Ind > EUR): p = {p_mw:.2e}')
print(f"Cohen's d (pooled): {d:.2f} → efecto medio")
print(f'n (indígenas) = {len(ind_froh)}, n (EUR) = {len(eur_froh)}')
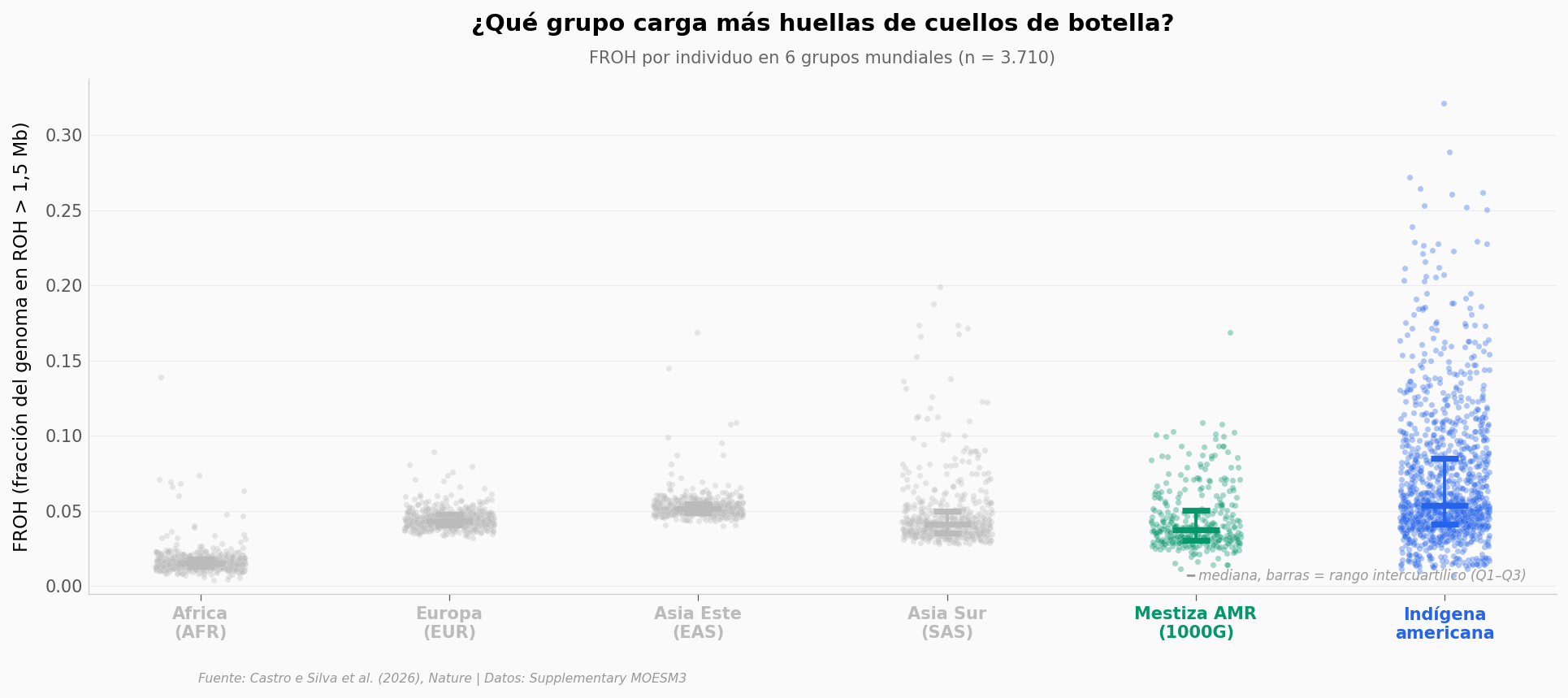
Mediana FROH — Indígena americana: 0.0535
Mediana FROH — Europea (EUR): 0.0436
Razón de medianas: 1.23x
Mann-Whitney U (Ind > EUR): p = 1.21e-32
Cohen's d (pooled): 0.64 → efecto medio
n (indígenas) = 1206, n (EUR) = 503
Lo que dicen los datos#
La mediana de FROH en los pueblos indígenas americanos está alrededor de 0,054 — aproximadamente 1,2 veces la mediana europea (0,044) y 3,5 veces la africana (0,015).
Todo apunta a que los antepasados de estos 45 pueblos pasaron por cuellos de botella más estrechos que la mayoría de las otras grandes poblaciones humanas. La diferencia con Europa es un efecto medio (Cohen’s d = 0,64), no dramático, pero muy consistente: el test Mann-Whitney da una p prácticamente cero (1,2 × 10⁻³²) sobre 1.709 individuos comparados.
Una nota de precaución: esto son datos del mismo panel de SNPs procesados con el mismo flujo, pero comparaciones de ROH entre estudios distintos son notoriamente sensibles al coverage y al filtrado. Con esa advertencia, el orden de magnitud es sólido — los pueblos indígenas americanos llegan a tener ROH por encima de EUR y cercanos al grupo de Asia Este (mediana 0,052).
El modelo simple de «una sola ola»#
Si los ancestros se movieron en una sola expansión continua hacia el sur, esperaríamos aislamiento por distancia: poblaciones más lejanas geográficamente deberían ser más distintas genéticamente.
Para medirlo usamos la distancia genética que reporta el paper (pairwise outgroup-f3) entre 1.378 pares de poblaciones indígenas. Si el modelo simple funciona, la nube de puntos debería caer en diagonal.
fig, ax = plt.subplots(figsize=(13, 5.5))
# Categorías con orden explícito
cats = [
('SA-SA', 'Sudamérica — Sudamérica', COLOR_DATOS, 35),
('NA-SA', 'Norteamérica — Sudamérica', COLOR_ALERTA, 35),
('NA-NA', 'Norteamérica — Norteamérica', COLOR_REFERENCIA, 70),
]
for cat, label, color, size in cats:
sub = dist[dist['Comparison'] == cat]
ax.scatter(sub['Distance_Km'], sub['Pairwise_outf3'],
color=color, s=size, alpha=0.5,
edgecolors='white', linewidths=0.3,
label=f'{label} (n = {len(sub)})', zorder=4)
rho_global, p_global = stats.spearmanr(dist['Distance_Km'], dist['Pairwise_outf3'])
ax.set_xlabel('Distancia geográfica (km)', fontsize=11)
ax.set_ylabel('Distancia genética (pairwise outgroup-f3)', fontsize=11)
ax.set_title('¿La geografía predice la genética?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03, f'1.378 pares de poblaciones · Spearman ρ global = {rho_global:.2f}',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.legend(fontsize=9, loc='lower right', framealpha=0.9)
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.tight_layout()
plt.savefig('figuras/ibd_scatter.png', dpi=200, bbox_inches='tight')
plt.show()
print(f'Global: ρ = {rho_global:+.3f} (p = {p_global:.2e}, n = {len(dist)})')
for cat, _, _, _ in cats:
sub = dist[dist['Comparison'] == cat]
if len(sub) >= 3:
rho, p = stats.spearmanr(sub['Distance_Km'], sub['Pairwise_outf3'])
print(f'{cat:6s} (n = {len(sub):4d}): ρ = {rho:+.3f} (p = {p:.2e})')
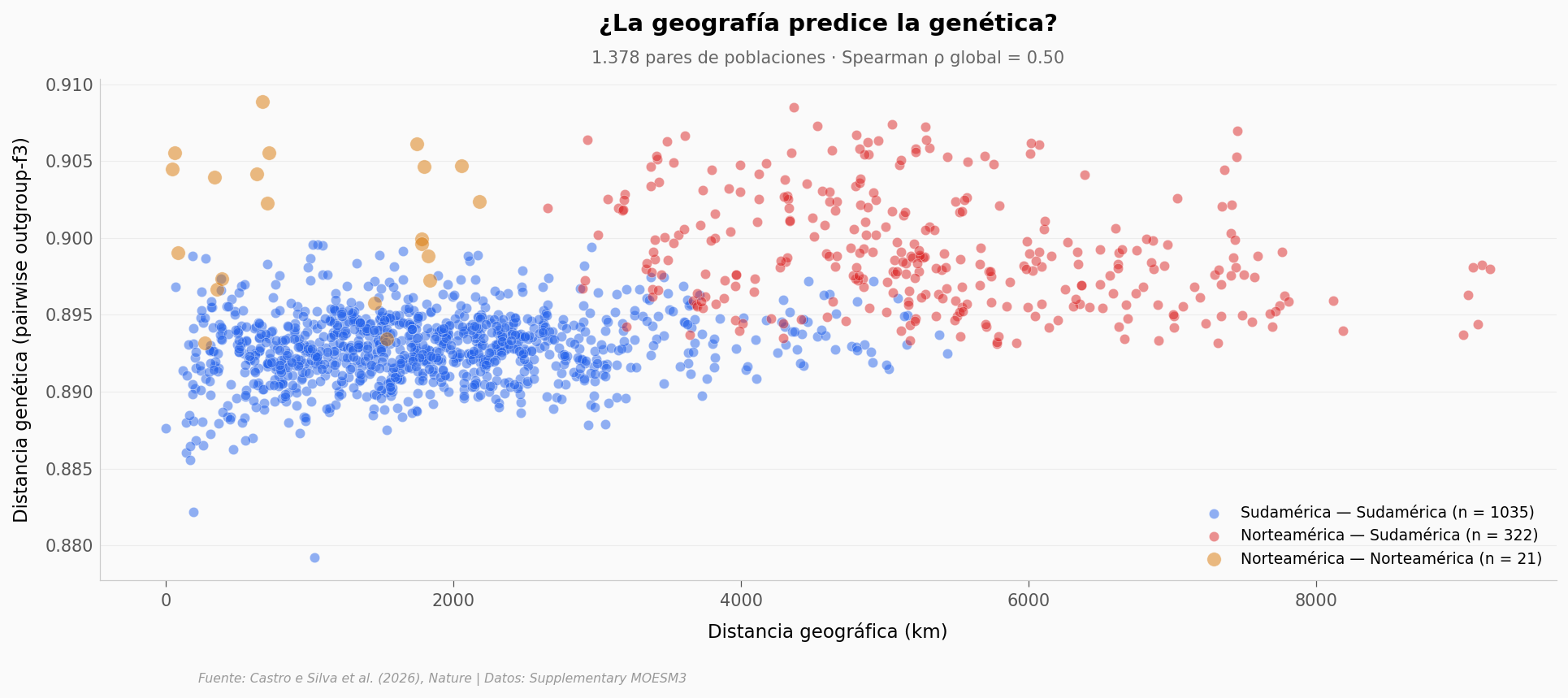
Global: ρ = +0.497 (p = 6.30e-87, n = 1378)
SA-SA (n = 1035): ρ = +0.148 (p = 1.73e-06)
NA-SA (n = 322): ρ = -0.286 (p = 1.81e-07)
NA-NA (n = 21): ρ = +0.014 (p = 9.51e-01)
La paradoja de Simpson, en vivo#
Leer este gráfico en capas cambia la conclusión:
Globalmente, la correlación existe: ρ = 0,50. Poblaciones más lejanas son más distintas genéticamente.
Dentro de Sudamérica (1.035 pares), la correlación es débil (ρ ≈ 0,15).
Entre Norte y Sudamérica (322 pares), la correlación es negativa (ρ ≈ −0,29) — poblaciones más alejadas tienden a ser menos genéticamente distintas de lo esperado.
El patrón global no es «aislamiento por distancia». Es una paradoja de Simpson: la correlación aparece porque al pegar las tres categorías, los pares NA-SA (lejos Y muy distintos) inflan la pendiente. Dentro de cada grupo, la geografía por sí sola explica poco.
Esto concuerda con la interpretación de los autores: no hubo una sola ola expansiva, sino al menos tres dispersiones que poblaron Sudamérica por rutas distintas.
La pista que nadie esperaba#
Desde 2015 se sabe que algunas poblaciones amazónicas comparten fragmentos de ADN con pueblos de Papúa Nueva Guinea, Australia y las islas Andamán. La llamaron la «ancestría Y» (o Ypykuéra).
Para cada población, contamos en cuántos tests estadísticos (\(D\)-statistic con \(|Z|>3\)) aparece esa señal australasiática — son los conteos que publicó el paper. Más tests significativos = huella más fuerte.
Compararon 36 poblaciones antiguas (muestras arqueológicas) contra 114 contemporáneas.
fig, ax = plt.subplots(figsize=(13, 5.5))
ancient_vals = austral['n_sig_ancient'].dropna().values
contemp_vals = austral['n_sig_contemporary'].dropna().values
np.random.seed(42)
positions = [0, 1.2]
data_pairs = [
(ancient_vals, COLOR_ALERTA, 'Poblaciones antiguas\n(n = 36)'),
(contemp_vals, COLOR_DATOS, 'Poblaciones contemporáneas\n(n = 114)'),
]
for i, (vals, color, _) in enumerate(data_pairs):
n = len(vals)
x_jitter = np.linspace(positions[i] - 0.15, positions[i] + 0.15, n)
np.random.shuffle(x_jitter)
ax.scatter(x_jitter, vals, color=color, s=40, alpha=0.55,
edgecolors='white', linewidths=0.4, zorder=4)
mediana = np.median(vals)
ax.errorbar(positions[i], mediana, yerr=None,
fmt='_', color=color, markersize=32, markeredgewidth=3.5, zorder=6)
top_ancient = austral.dropna(subset=['n_sig_ancient']).nlargest(3, 'n_sig_ancient')
for _, row in top_ancient.iterrows():
name = row['ancient_pop'].replace('_', ' ')[:28]
ax.annotate(name, xy=(positions[0] + 0.22, row['n_sig_ancient']),
fontsize=8.5, color=COLOR_ALERTA, va='center')
ax.set_xticks(positions)
ax.set_xticklabels([p[2] for p in data_pairs], fontsize=10, fontweight='bold')
for tick, color in zip(ax.get_xticklabels(), [COLOR_ALERTA, COLOR_DATOS]):
tick.set_color(color)
ax.set_ylabel('Nº de tests D con |Z| > 3 (señal australasiática)', fontsize=11)
ax.set_title('¿Dónde se concentra la huella australasiática?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03, 'Mayor valor = más pruebas de ancestría compartida con Papúa/Australia',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
ax.text(0.98, 0.02, '━ mediana · cada punto = una población',
transform=ax.transAxes, fontsize=8, color='#999999',
ha='right', va='bottom', style='italic')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.tight_layout()
plt.savefig('figuras/australasia_compare.png', dpi=200, bbox_inches='tight')
plt.show()
u, p_mw = stats.mannwhitneyu(ancient_vals, contemp_vals, alternative='two-sided')
pooled_sd = np.sqrt(((len(ancient_vals) - 1) * ancient_vals.var(ddof=1) + (len(contemp_vals) - 1) * contemp_vals.var(ddof=1)) / (len(ancient_vals) + len(contemp_vals) - 2))
d = (ancient_vals.mean() - contemp_vals.mean()) / pooled_sd
print(f'Mediana antiguas: {np.median(ancient_vals):.1f} (máx: {ancient_vals.max():.0f})')
print(f'Mediana contemporáneas: {np.median(contemp_vals):.1f} (máx: {contemp_vals.max():.0f})')
print(f'Mann-Whitney U (2 colas): p = {p_mw:.3f}')
print(f"Cohen's d (pooled): {d:.2f} → efecto grande")
print()
print('Top 3 antiguas (llevan la cola):')
for _, r in top_ancient.iterrows():
print(f" {r['ancient_pop']:36s} {int(r['n_sig_ancient']):>4d} tests")
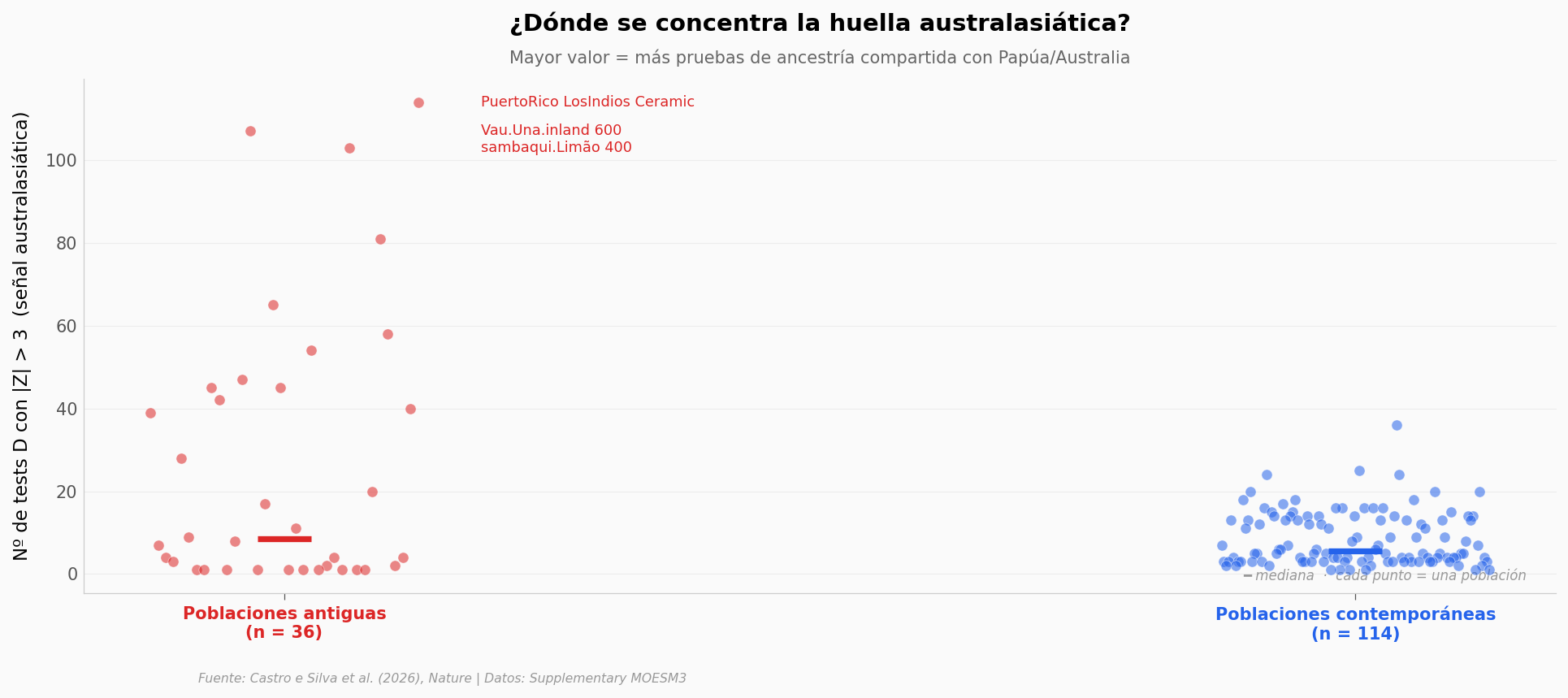
Mediana antiguas: 8.5 (máx: 114)
Mediana contemporáneas: 5.5 (máx: 36)
Mann-Whitney U (2 colas): p = 0.341
Cohen's d (pooled): 1.07 → efecto grande
Top 3 antiguas (llevan la cola):
PuertoRico_LosIndios_Ceramic 114 tests
Vau.Una.inland_600 107 tests
sambaqui.Limão_400 103 tests
¿Qué tan raro es este patrón?#
El paper dice que unas pocas poblaciones antiguas llevan la señal Ypykuéra a una intensidad muy por encima del resto. Veamos la distribución completa y dónde cae cada dato contemporáneo.
fig, ax = plt.subplots(figsize=(12, 5))
n_vals, bins, _ = ax.hist(ancient_vals, bins=18, color=COLOR_ALERTA, alpha=0.35,
edgecolor=COLOR_ALERTA, linewidth=0.8, label='Poblaciones antiguas')
y_max = n_vals.max() * 1.25
ax.set_ylim(0, y_max)
mediana_ancient = np.median(ancient_vals)
mediana_contemp = np.median(contemp_vals)
ax.axvline(x=mediana_ancient, color=COLOR_ALERTA, linewidth=1.5, linestyle='--', alpha=0.8)
ax.axvline(x=mediana_contemp, color=COLOR_DATOS, linewidth=2.5)
ax.text(mediana_contemp + 1.5, y_max * 0.92,
f'mediana contemporáneas\n= {mediana_contemp:.0f}',
fontsize=9.5, color=COLOR_DATOS, fontweight='bold')
ax.text(mediana_ancient + 1.5, y_max * 0.72,
f'mediana antiguas\n= {mediana_ancient:.0f}',
fontsize=9.5, color=COLOR_ALERTA, fontweight='bold')
top3 = sorted(ancient_vals, reverse=True)[:3]
for v in top3:
ax.annotate(f'{int(v)}', xy=(v, 0.3),
xytext=(v, y_max * 0.45),
fontsize=9, color=COLOR_ALERTA, ha='center',
arrowprops=dict(arrowstyle='->', color=COLOR_ALERTA, lw=1.2, alpha=0.6))
ax.set_xlabel('Nº de tests D con |Z| > 3', fontsize=11)
ax.set_ylabel('Nº de poblaciones antiguas', fontsize=11)
ax.set_title('La cola larga: unas pocas poblaciones antiguas llevan casi toda la señal',
fontsize=13, fontweight='bold', pad=28)
ax.text(0.5, 1.03, 'Distribución de señal australasiática — 36 poblaciones antiguas',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.tight_layout()
plt.savefig('figuras/australasia_histograma.png', dpi=200, bbox_inches='tight')
plt.show()
max_contemp = contemp_vals.max()
n_above = (ancient_vals > max_contemp).sum()
print(f'Máximo en 114 poblaciones contemporáneas: {int(max_contemp)} tests')
print(f'Antiguas por encima de ese máximo: {n_above} de 36 ({100*n_above/36:.0f}%)')
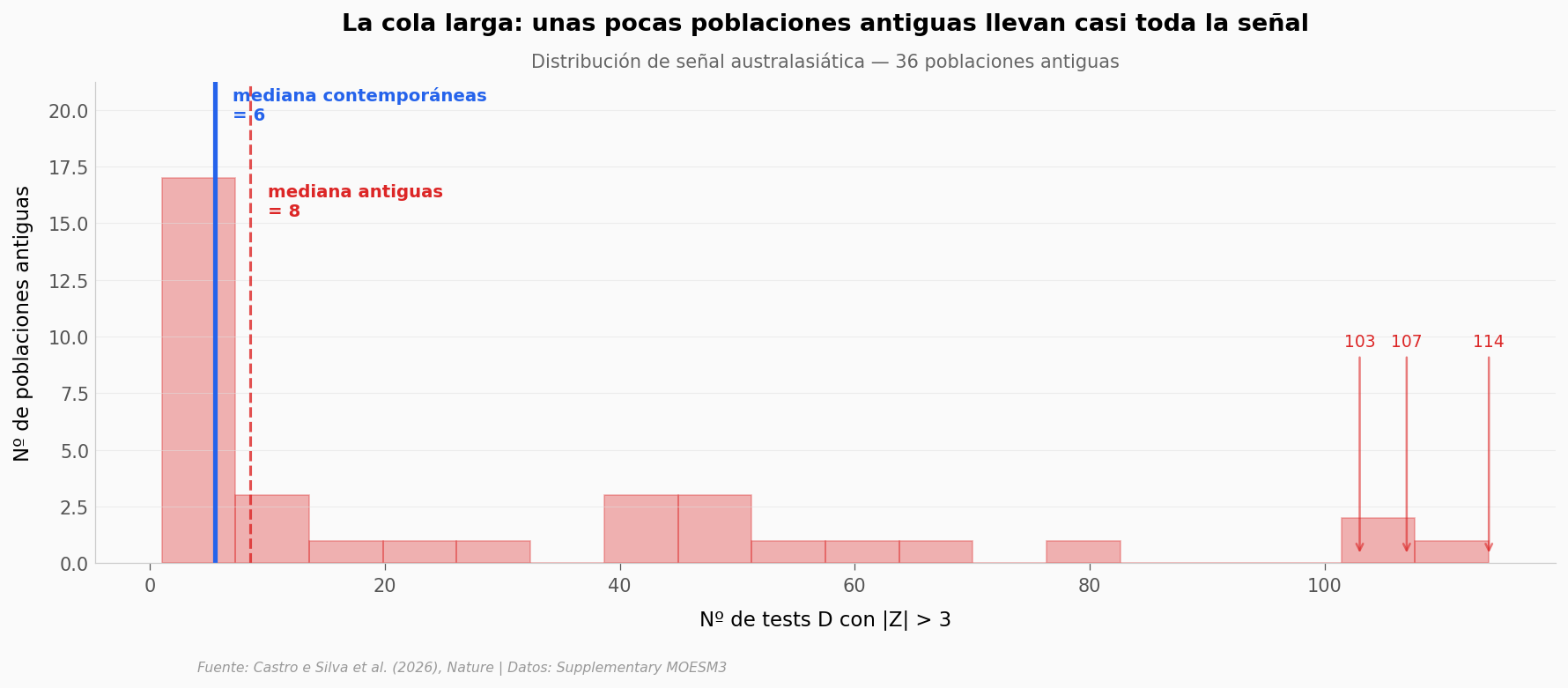
Máximo en 114 poblaciones contemporáneas: 36 tests
Antiguas por encima de ese máximo: 13 de 36 (36%)
Lo que los datos soportan#
Afirmación |
¿Soportada? |
Detalle |
|---|---|---|
128 genomas nuevos, 45 poblaciones, 8 países |
✅ |
Tabla 1 lo confirma cuenta por cuenta (ver celda 2) |
Los pueblos indígenas americanos tienen FROH más alto que europeos |
✅ |
Mediana 0,054 vs 0,044 (1,2×). d = 0,64 (medio), p = 1,2 × 10⁻³². Mismo panel de SNPs |
Aislamiento por distancia dentro de América |
⚠️ |
Global ρ = 0,50, pero dentro de Sudamérica ρ = 0,15 y entre Norte y Sudamérica ρ = −0,29. El patrón agregado se debe a la mezcla de categorías (paradoja de Simpson) |
Las poblaciones antiguas muestran mayor señal australasiática que las contemporáneas |
⚠️ |
d = 1,07 (grande) pero Mann-Whitney p = 0,34. El efecto lo llevan pocos outliers antiguos (sambaqui, Puerto Rico Ceramic). El paper lo enmarca como patrón descriptivo, no como diferencia promedio |
La señal australasiática existe en muestras de ~6.800 años |
✅ |
Argentina_LagunaChica_6800BP aparece con 81 tests Z > 3 — consistente con que la huella es pre-colombina |
Limitaciones: (1) estos son datos agregados que publicó el paper; los genomas individuales están en EGA bajo acceso controlado. (2) Las estimaciones de ROH son sensibles al coverage y al filtrado, aunque aquí los autores usaron el mismo flujo para todos los individuos. (3) La comparación antiguas vs contemporáneas en la señal australasiática tiene tamaños de muestra asimétricos (36 vs 114) y varianza desigual — por eso el p es alto pese al d grande.
Ahora tú#
Tres cosas que puedes explorar con una línea de código:
¿Qué familia lingüística une a los pueblos más distantes geográficamente? Pista:
genomas.groupby('Language').agg(paises=('Country', 'nunique')).¿Los pueblos del Chaco tienen menos o más ROH que los del Western_South_America? Pista: cruza
genomas['Clusters']con los IID deroh.¿La señal australasiática correlaciona con la latitud de la población antigua? Las poblaciones antiguas están en la columna
ancient_popdeaustral; mapea a las coordenadas degenomascuando los nombres coincidan.
# --- EXPERIMENTA AQUÍ ---
# Pregunta: ¿los pueblos del Chaco cargan más o menos FROH que los del Western South America?
# 1. Cruzar tabla de genomas (con Clusters) con tabla de ROH (por IID)
cluster_map = genomas.set_index('IID')['Clusters'].to_dict()
roh_new = roh[roh['IID'].isin(cluster_map)].copy()
roh_new['Cluster'] = roh_new['IID'].map(cluster_map)
# 2. Comparar medianas de FROH por cluster geográfico
resumen = roh_new.groupby('Cluster').agg(
n=('FROH', 'size'),
mediana_FROH=('FROH', 'median'),
media_FROH=('FROH', 'mean'),
).round(4).sort_values('mediana_FROH', ascending=False)
print(resumen)
# ¿Cómo cambia esto si miras solo poblaciones con N >= 10? Prueba tú.
n mediana_FROH media_FROH
Cluster
Unclear 4 0.1978 0.1886
Western_South_America 48 0.1213 0.1330
Chaco 13 0.1091 0.1335
Eastern_South_America 47 0.1079 0.1176
Southern_Cone 6 0.0877 0.0856
Aridoamerica 4 0.0733 0.0755
Mesoamerica 6 0.0720 0.0755
Fuentes#
Paper: The evolutionary history and unique genetic diversity of Indigenous Americans
Nature, 2026-04-22
Datos: Supplementary Tables 1–23 (MOESM3) — metadata, ROH, genetic/geographic distances, Australasian D-statistics
Springer Nature Supplementary, 2026-04-22
13 afirmaciones verificadas contra estas fuentes
Repositorio del notebook: Ciencia-a-Mordiscos/lab Licencia notebook: CC-BY 4.0 · Datos: publicados por los autores bajo licencia de Springer Nature Canal: Ciencia a Mordiscos — divulgación científica con datos verificables