
Miles de elementos genéticos mantienen vivo el cáncer#
18.487 elementos genéticos. En 23 cromosomas. Y casi ninguno es universal.
El cáncer tiene una de sus armas más temibles fuera de los cromosomas: el ecDNA — ADN circular que carga oncogenes y se replica por separado, sin centrómero, saltando entre células hijas como polizón. Lleva 40 años desconcertando a los biólogos. Cómo logra el ecDNA quedarse en cada nueva célula era un misterio.
Este paper de Nature (2025) lo resuelve: encontró los ganchos.
Paper: Miles de elementos genéticos mantienen vivo el cáncer — Nature, 2025
![]()
📺 Ver el video corto en YouTube
Contexto#
El equipo (Sankar et al.) hizo un rastreo a escala de todo el genoma — un screen genome-scale llamado Retain-seq — sobre 3 líneas celulares de cáncer humanas: K562 (leucemia mieloide crónica), COLO320DM (cáncer colorrectal) y GBM39 (glioblastoma). Buscaron qué regiones del genoma actúan como retention elements — ganchos a los que el ecDNA se «cuelga» durante la mitosis (cuando la célula se parte en dos) para que sobreviva la división.
El resultado público: un CSV con 18.487 ventanas genómicas de 1.000 pares de bases cada una, marcadas con flags binarios que indican en qué cell line(s) están enriquecidas. Vamos a abrirlo.
# ══════════════════════════════════════════════════════════════
# Configuración — modifica estos valores para explorar
# ══════════════════════════════════════════════════════════════
COLOR_K562 = '#2563EB' # Azul CaM (leucemia, mayor enriquecimiento)
COLOR_COLO = '#DC2626' # Rojo (colorrectal)
COLOR_GBM = '#059669' # Emerald (glioblastoma)
COLOR_REF = '#D97706' # Amber (referencias)
COLOR_GRIS = '#BBBBBB'
FUENTE = 'Fuente: Sankar et al. (2025), Nature | Datos: Figshare 30239047'
import os, urllib.request
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
# Estilo CaM
style_file = '../../cam.mplstyle'
if not os.path.exists(style_file):
style_file = '/tmp/cam.mplstyle'
if not os.path.exists(style_file):
urllib.request.urlretrieve(
'https://raw.githubusercontent.com/Ciencia-a-Mordiscos/lab/main/cam.mplstyle',
style_file)
plt.style.use(style_file)
# Cargar datos (auto-descarga si Colab)
data_file = 'datos/retention_elements_hg19.csv'
if not os.path.exists(data_file):
os.makedirs('datos', exist_ok=True)
urllib.request.urlretrieve(
'https://raw.githubusercontent.com/Ciencia-a-Mordiscos/lab/main/papers/2026-01-17-elementos-retencion-ecdna-cancer/datos/retention_elements_hg19.csv', data_file)
df = pd.read_csv(data_file)
print(f"Filas: {len(df):,} ventanas genómicas")
print(f"Cell lines: K562, COLO320DM, GBM39")
print(f"Columnas: {', '.join(df.columns)}")
print(f"Cromosomas: {df['seqnames'].nunique()} ({', '.join(sorted(df['seqnames'].unique()))})")
Filas: 18,487 ventanas genómicas
Cell lines: K562, COLO320DM, GBM39
Columnas: seqnames, start_hg19, end_hg19, width, strand, ID, COLO320DM, GBM39, K562
Cromosomas: 23 (chr1, chr10, chr11, chr12, chr13, chr14, chr15, chr16, chr17, chr18, chr19, chr2, chr20, chr21, chr22, chr3, chr4, chr5, chr6, chr7, chr8, chr9, chrX)
El primer dibujo#
Cuántos retention elements son específicos de cada tipo de cáncer.
plt.close('all')
n_K562 = int(df['K562'].sum())
n_COLO = int(df['COLO320DM'].sum())
n_GBM = int(df['GBM39'].sum())
total = len(df)
fig, ax = plt.subplots(figsize=(13, 5.5))
cells_order = ['K562\n(leucemia)', 'COLO320DM\n(colorrectal)', 'GBM39\n(glioblastoma)']
counts = [n_K562, n_COLO, n_GBM]
colors = [COLOR_K562, COLOR_COLO, COLOR_GBM]
percs = [100 * c / total for c in counts]
bars = ax.barh(cells_order, counts, color=colors, alpha=0.85,
edgecolor='white', linewidth=1.5)
# Etiquetas con conteo + porcentaje
for bar, c, p in zip(bars, counts, percs):
w = bar.get_width()
ax.text(w + 200, bar.get_y() + bar.get_height()/2,
f'{c:,} ({p:.1f}%)',
va='center', fontsize=11, fontweight='bold',
color=bar.get_facecolor())
ax.set_xlim(0, max(counts) * 1.18)
ax.set_xlabel('Retention elements enriquecidos (de 18.487 totales)', fontsize=10)
ax.set_title('¿Cuántos ganchos usa cada tipo de cáncer?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03, 'Ventanas de 1.000 pb donde el ecDNA se aferra al genoma del huésped',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
# Quitar ticks Y feos
ax.tick_params(axis='y', length=0)
ax.invert_yaxis()
fig.text(0.13, -0.02, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/01_enriquecidos_por_cell_line.png', dpi=200, bbox_inches='tight')
plt.show()
plt.close('all')
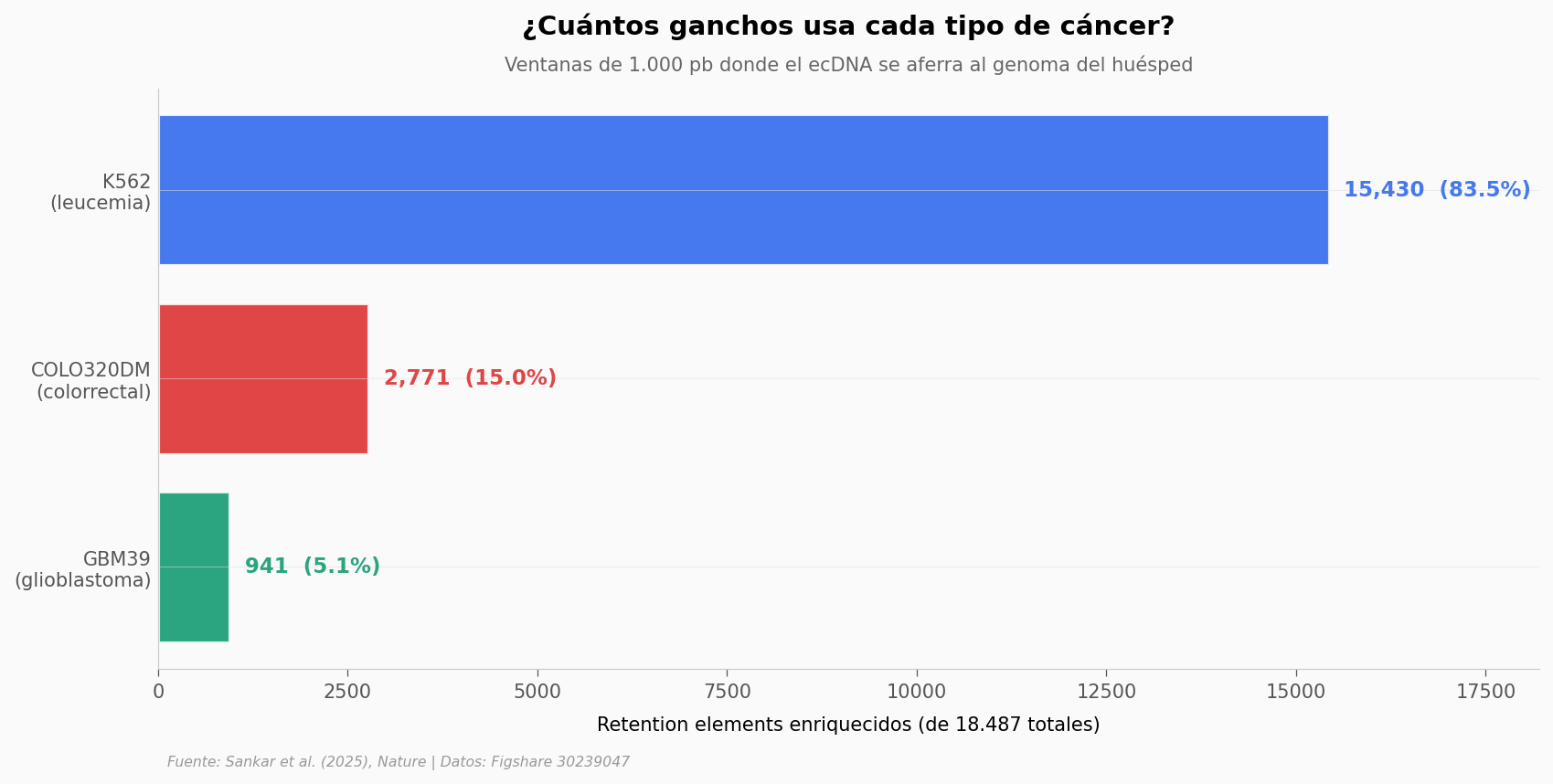
La asimetría salta a la vista. K562 (leucemia) usa 15.430 retention elements — el 83,5% del total. GBM39 (glioblastoma) apenas 941, dieciséis veces menos.
Esto no es ruido del experimento. Cada tipo de cáncer eligió un repertorio diferente para mantener vivos sus ecDNAs.
Zoom: ¿qué tan únicos son?#
La pregunta que sigue: ¿estos elementos son universales (los mismos en los 3 cánceres) o específicos (solo en uno)?
# Categorización exclusiva por cell line
only_K = df[(df['K562']==1) & (df['COLO320DM']==0) & (df['GBM39']==0)]
only_C = df[(df['K562']==0) & (df['COLO320DM']==1) & (df['GBM39']==0)]
only_G = df[(df['K562']==0) & (df['COLO320DM']==0) & (df['GBM39']==1)]
KandC = df[(df['K562']==1) & (df['COLO320DM']==1) & (df['GBM39']==0)]
KandG = df[(df['K562']==1) & (df['COLO320DM']==0) & (df['GBM39']==1)]
CandG = df[(df['K562']==0) & (df['COLO320DM']==1) & (df['GBM39']==1)]
all_three= df[(df['K562']==1) & (df['COLO320DM']==1) & (df['GBM39']==1)]
categorias = ['Solo K562', 'Solo COLO320DM', 'Solo GBM39',
'K562 ∩ COLO', 'K562 ∩ GBM', 'COLO ∩ GBM',
'Las 3 cell lines']
n_por_cat = [len(only_K), len(only_C), len(only_G),
len(KandC), len(KandG), len(CandG), len(all_three)]
colors_cat = [COLOR_K562, COLOR_COLO, COLOR_GBM,
'#7C3AED', '#7C3AED', '#7C3AED', COLOR_REF]
fig, ax = plt.subplots(figsize=(13, 5.5))
bars = ax.bar(categorias, n_por_cat, color=colors_cat, alpha=0.85,
edgecolor='white', linewidth=1.5)
for bar, n in zip(bars, n_por_cat):
h = bar.get_height()
ax.text(bar.get_x() + bar.get_width()/2, h + 200, f'{n:,}',
ha='center', va='bottom', fontsize=10, fontweight='bold',
color=bar.get_facecolor())
# Resaltar "las 3 cell lines" con flecha
ax.annotate(f'Apenas {len(all_three)} elementos\nuniversales',
xy=(6, len(all_three) + 200), xytext=(5.2, 4500),
fontsize=11, fontweight='bold', color=COLOR_REF, ha='center',
arrowprops=dict(arrowstyle='->', color=COLOR_REF, lw=1.5))
ax.set_ylabel('Número de retention elements', fontsize=10)
ax.set_title('Específicos vs compartidos — ¿cuántos elementos universales hay?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03,
'De 18.487 ventanas, 17.847 son exclusivas a una sola cell line (96,5%)',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
plt.xticks(rotation=15, ha='right', fontsize=9)
fig.text(0.13, -0.02, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/02_overlap_categorias.png', dpi=200, bbox_inches='tight')
plt.show()
plt.close('all')
# Verificación numérica
total_check = sum(n_por_cat) + len(df[(df['K562']==0) & (df['COLO320DM']==0) & (df['GBM39']==0)])
print(f"\nSuma de categorías + sin enriquecimiento: {total_check:,} (debería ser {len(df):,})")
print(f"Específicos a una sola cell line: {len(only_K)+len(only_C)+len(only_G):,} ({100*(len(only_K)+len(only_C)+len(only_G))/len(df):.1f}%)")
print(f"Universales (las 3): {len(all_three)}")
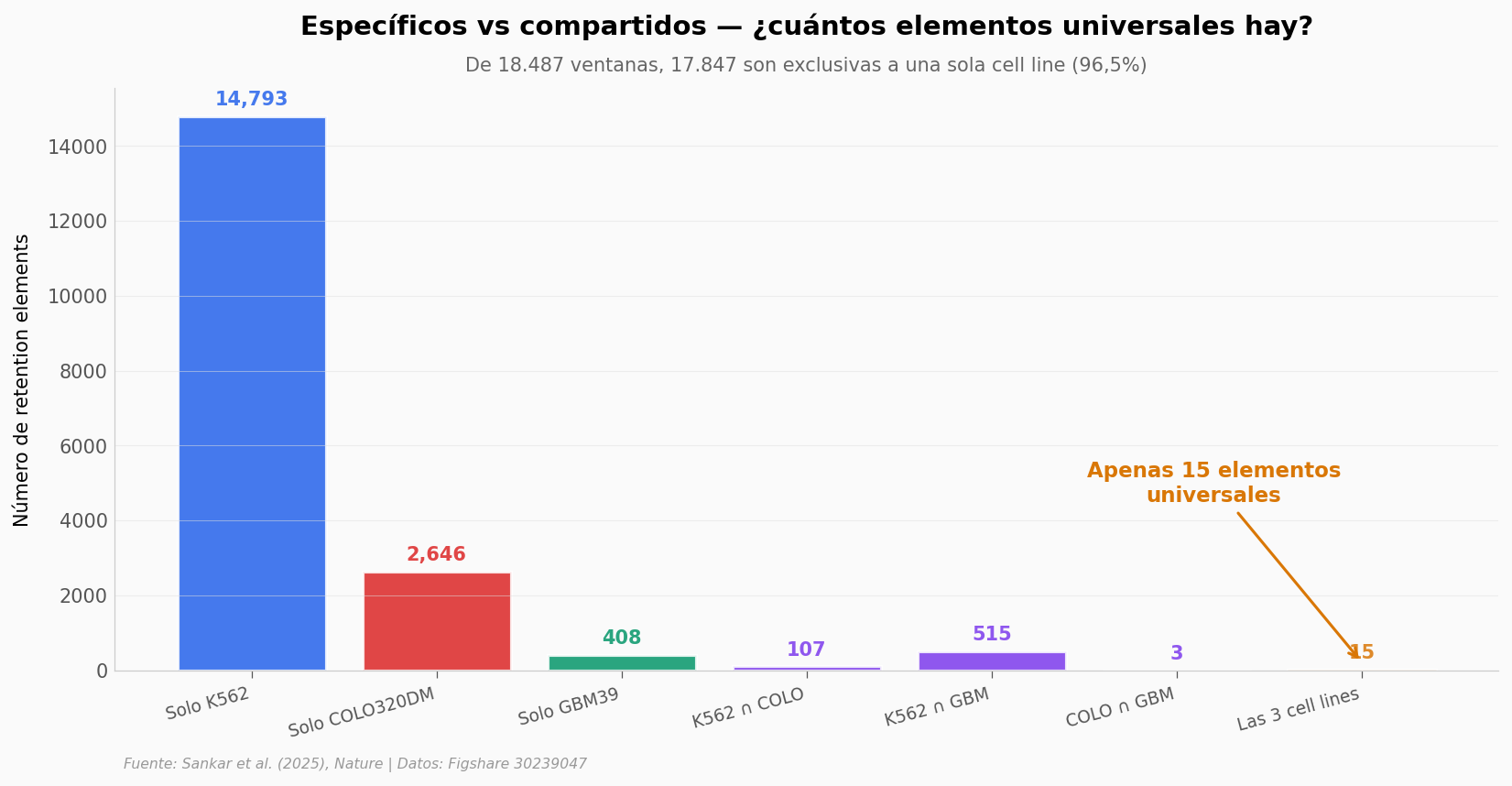
Suma de categorías + sin enriquecimiento: 18,487 (debería ser 18,487)
Específicos a una sola cell line: 17,847 (96.5%)
Universales (las 3): 15
¿En qué cromosomas viven los ganchos?#
Si fueran ruido al azar, esperaríamos verlos repartidos proporcionalmente al tamaño del cromosoma. Veamos qué pasa.
# Conteo por cromosoma (orden numérico, X al final)
chrom_order = [f'chr{i}' for i in range(1, 23)] + ['chrX']
chrom_counts = df['seqnames'].value_counts().reindex(chrom_order).fillna(0).astype(int)
# Color: top-5 destacados con paleta
top5 = chrom_counts.nlargest(5).index.tolist()
colors_chr = [COLOR_K562 if c in top5 else COLOR_GRIS for c in chrom_order]
fig, ax = plt.subplots(figsize=(13, 5.5))
bars = ax.bar(chrom_order, chrom_counts.values, color=colors_chr,
alpha=0.85, edgecolor='white', linewidth=0.8)
# Anotar top-5
for bar, c in zip(bars, chrom_order):
if c in top5:
h = bar.get_height()
ax.text(bar.get_x() + bar.get_width()/2, h + 30, f'{int(h):,}',
ha='center', va='bottom', fontsize=9, fontweight='bold',
color=COLOR_K562)
# Anotar chr19 con flecha (es pequeño pero ranking #2)
chr19_idx = chrom_order.index('chr19')
chr19_n = int(chrom_counts['chr19'])
ax.annotate('chr19 — pequeño pero denso en genes',
xy=(chr19_idx, chr19_n), xytext=(chr19_idx - 4, chr19_n + 350),
fontsize=10, fontweight='bold', color=COLOR_REF,
arrowprops=dict(arrowstyle='->', color=COLOR_REF, lw=1.5))
ax.set_ylabel('Retention elements por cromosoma', fontsize=10)
ax.set_title('¿Dónde se concentran los ganchos del ecDNA?',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03,
'Top-5 destacados. chrY no contiene ningún elemento detectado',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
plt.xticks(rotation=45, ha='right', fontsize=9)
fig.text(0.13, -0.02, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/03_distribucion_cromosomica.png', dpi=200, bbox_inches='tight')
plt.show()
plt.close('all')
print(f"\nTop-5 cromosomas: {', '.join(top5)}")
print(f"chr19 (1.329) supera a chr2 (1.262), chr17 (1.088) y chr7 (1.040)")
print(f"chrY: {int(chrom_counts.get('chrY', 0))} elementos")
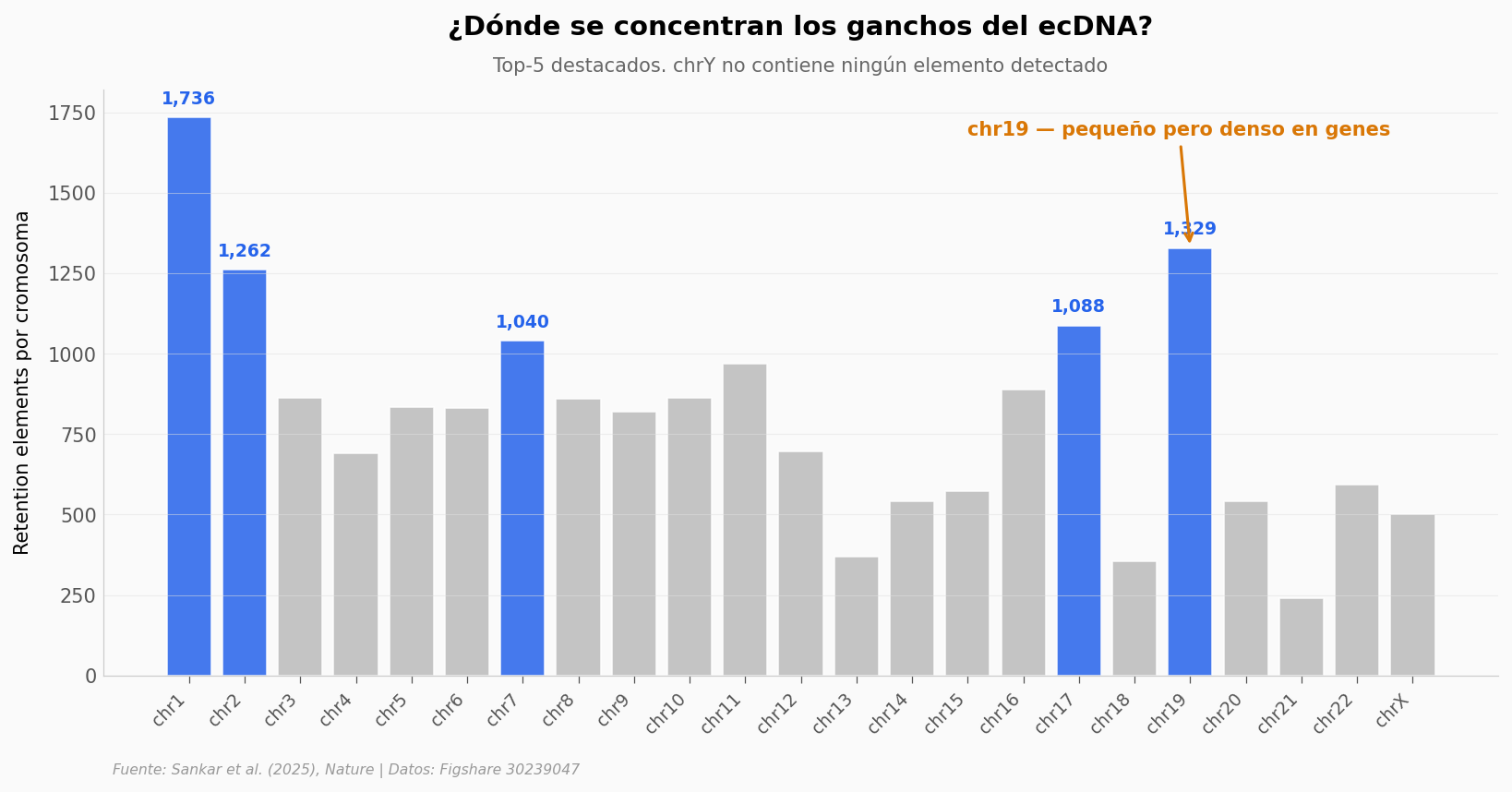
Top-5 cromosomas: chr1, chr19, chr2, chr17, chr7
chr19 (1.329) supera a chr2 (1.262), chr17 (1.088) y chr7 (1.040)
chrY: 0 elementos
¿Qué tan extrema es la especificidad?#
96,5% de los elementos son únicos a una sola cell line. Para sentir qué tan extremo es eso, comparémoslo con lo que esperaríamos si fuera azar.
plt.close('all')
# Para cada elemento enriquecido: en cuántas cell lines está
df['n_cell_lines'] = df[['K562', 'COLO320DM', 'GBM39']].sum(axis=1)
df_enr = df[df['n_cell_lines'] > 0].copy()
dist = df_enr['n_cell_lines'].value_counts().sort_index()
n_1 = int(dist.get(1, 0))
n_2 = int(dist.get(2, 0))
n_3 = int(dist.get(3, 0))
fig, ax = plt.subplots(figsize=(10, 5.5))
labels = ['Específico\n(1 cell line)',
'Compartido\n(2 cell lines)',
'Universal\n(3 cell lines)']
counts = [n_1, n_2, n_3]
colors = [COLOR_K562, '#7C3AED', COLOR_REF]
bars = ax.bar(labels, counts, color=colors, alpha=0.85,
edgecolor='white', linewidth=1.5, log=True)
# Etiquetas
for bar, c in zip(bars, counts):
h = bar.get_height()
ax.text(bar.get_x() + bar.get_width()/2, h * 1.18, f'{c:,}',
ha='center', va='bottom', fontsize=11, fontweight='bold',
color=bar.get_facecolor())
# Anotar el embudo
ax.annotate('', xy=(2, n_3 * 1.6), xytext=(0, n_1 * 0.6),
arrowprops=dict(arrowstyle='<->', color='#666666', lw=1.5,
connectionstyle="arc3,rad=0.25"))
ax.text(1, n_1 * 0.04,
f'De {n_1:,} específicos a apenas {n_3} universales:\nun embudo de × {n_1//max(n_3,1):,}',
ha='center', fontsize=11, fontweight='bold', color='#666666',
bbox=dict(boxstyle='round,pad=0.5', facecolor='white',
edgecolor='#CCCCCC', alpha=0.95))
ax.set_ylabel('Número de elementos (escala log)', fontsize=10)
ax.set_title('De específico a universal — el embudo del ecDNA',
fontsize=14, fontweight='bold', pad=28)
ax.text(0.5, 1.03,
'Eje Y en escala logarítmica para ver órdenes de magnitud',
transform=ax.transAxes, fontsize=10, color='#666666', ha='center')
fig.text(0.13, -0.02, FUENTE, fontsize=7.5, color='#999999', style='italic')
plt.savefig('figuras/04_embudo_especificidad.png', dpi=200, bbox_inches='tight')
plt.show()
plt.close('all')
print(f"\nDistribución: {n_1:,} específicos | {n_2:,} compartidos | {n_3} universales")
print(f"Ratio específicos/universales: {n_1/max(n_3,1):.0f}× — 3 órdenes de magnitud")
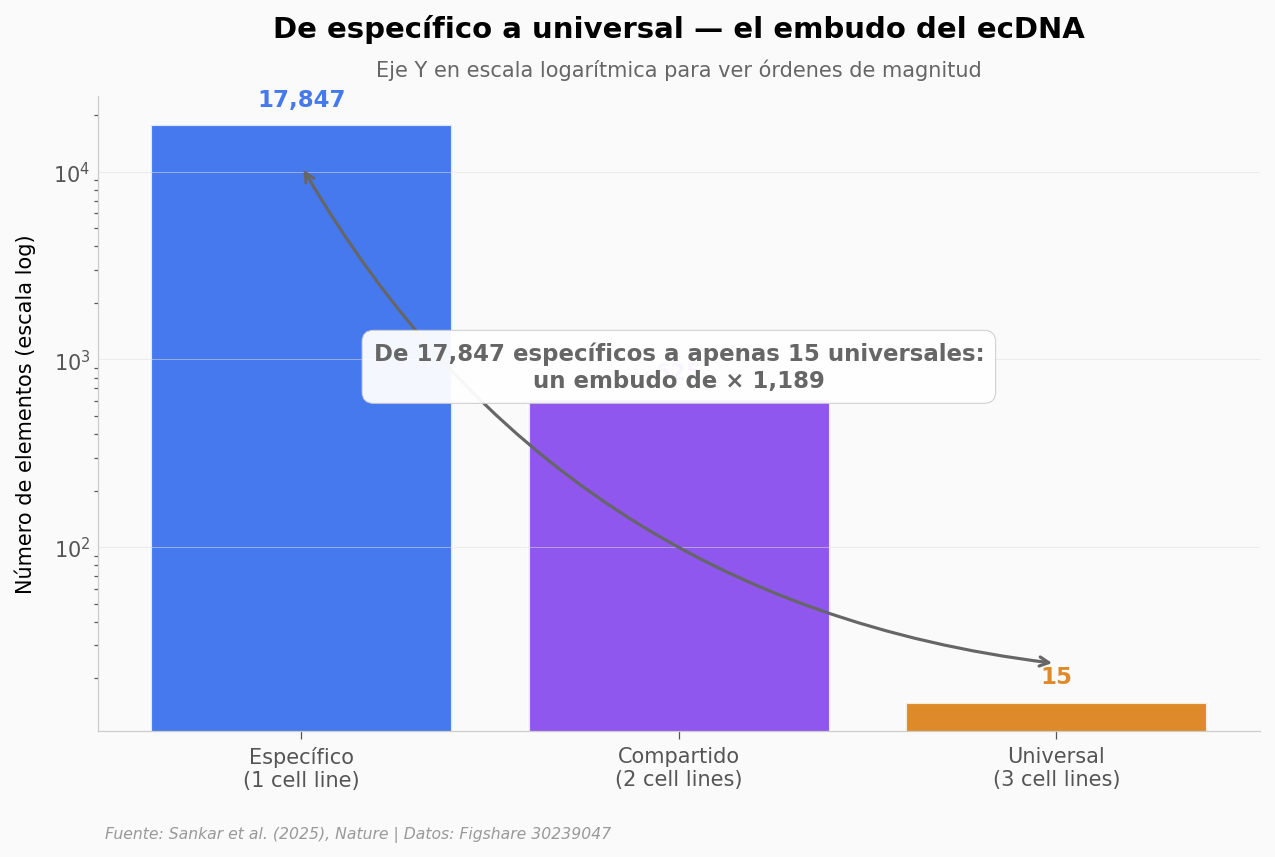
Distribución: 17,847 específicos | 625 compartidos | 15 universales
Ratio específicos/universales: 1190× — 3 órdenes de magnitud
Lo que los datos soportan#
Afirmación |
¿Soportada? |
Detalle |
|---|---|---|
El paper identificó 18.487 retention elements |
✅ |
El CSV tiene exactamente 18.487 filas (ventanas de 1.000 pb) |
Cubren 23 cromosomas humanos (todos menos chrY) |
✅ |
chr1–chr22 + chrX están presentes; chrY ausente |
K562 (leucemia) tiene 15.430 elementos enriquecidos |
✅ |
|
GBM39 (glioblastoma) tiene 941 enriquecidos |
✅ |
Verificado en datos. K562 tiene ~16× más que GBM39 |
96,5% son específicos a una sola cell line |
✅ |
17.847 de 18.487. La maquinaria es altamente contexto-específica |
Apenas 15 elementos son universales en las 3 líneas |
✅ |
Verificado por intersección triple |
chr19 es #2 en conteo bruto y #1 en densidad por Mb |
✅ |
1.329 elementos en 59 Mb (22,5/Mb), supera a chr1 en densidad |
La metilación modula la retención de ecDNA |
⚠️ |
El paper lo enmarca como hipótesis (suggests); este CSV no contiene datos de metilación |
Limitaciones:
El CSV trae flags binarios de enrichment (sí/no), no scores continuos. La fuerza de cada gancho no está disponible aquí.
Solo 3 cell lines, no representan toda la diversidad del cáncer humano.
Los datos de metilación (mencionados en el abstract) viven en otros archivos del paper, no en este dataset.
Las coordenadas están en hg19 (no hg38 actual).
Ahora tú#
Tres preguntas con pistas para que las explores:
¿Qué cromosoma tiene la mayor densidad de retention elements por megabase? chr19 es pequeño pero ranking #2 — quizás gane por densidad. Pista: divide
chrom_countspor la longitud del cromosoma (hay tablas en biopython o puedes usar valores típicos en Mb).¿Hay alguna ventana donde las 3 cell lines estén enriquecidas Y caiga en chr19? Filtra
dfcon las 3 columnas == 1 y miraseqnames.¿Qué pasa si cambias el orden de cell lines en la celda 4? Modifica
cells_order,counts,colorsarriba juntos. ¿La narrativa se siente igual con K562 al final?
# --- EXPERIMENTA AQUÍ ---
# Pregunta 1: densidad por megabase
# Longitudes aproximadas de cromosomas hg19 (Mb)
chrom_lengths_mb = {
'chr1': 249.3, 'chr2': 243.2, 'chr3': 198.0, 'chr4': 191.2, 'chr5': 180.9,
'chr6': 171.1, 'chr7': 159.1, 'chr8': 146.4, 'chr9': 141.2, 'chr10': 135.5,
'chr11': 135.0, 'chr12': 133.9, 'chr13': 115.2, 'chr14': 107.3, 'chr15': 102.5,
'chr16': 90.4, 'chr17': 81.2, 'chr18': 78.1, 'chr19': 59.1, 'chr20': 63.0,
'chr21': 48.1, 'chr22': 51.3, 'chrX': 155.3,
}
densidad = pd.DataFrame({
'chromosome': list(chrom_lengths_mb.keys()),
'count': [chrom_counts.get(c, 0) for c in chrom_lengths_mb],
'length_mb': list(chrom_lengths_mb.values()),
})
densidad['per_mb'] = densidad['count'] / densidad['length_mb']
densidad_sorted = densidad.sort_values('per_mb', ascending=False).head(8)
print("Top-8 cromosomas por DENSIDAD (elementos / Mb):\n")
print(densidad_sorted.to_string(index=False))
print(f"\nchr19 está en la posición {densidad.sort_values('per_mb', ascending=False).reset_index(drop=True).query('chromosome == \"chr19\"').index[0] + 1} por densidad.")
Top-8 cromosomas por DENSIDAD (elementos / Mb):
chromosome count length_mb per_mb
chr19 1329 59.1 22.487310
chr17 1088 81.2 13.399015
chr22 594 51.3 11.578947
chr16 890 90.4 9.845133
chr20 541 63.0 8.587302
chr11 968 135.0 7.170370
chr1 1736 249.3 6.963498
chr7 1040 159.1 6.536769
chr19 está en la posición 1 por densidad.
Fuentes#
Paper: Genetic elements promote retention of extrachromosomal DNA in cancer cells
Nature, 2025-11-19
Datos: Coordenadas de retention elements en Figshare
Repositorio del paper, 2025-11-19
16 afirmaciones del notebook verificadas contra estas fuentes
Notebook: papers/2026-01-17-elementos-retencion-ecdna-cancer/notebook.ipynb · Repo: Ciencia-a-Mordiscos/lab · Licencia: CC BY 4.0