
Una sola metilación apaga la enzima#
Una marca química diminuta — un grupo metilo en el carbono 5 de una citosina del ADN — basta para que la herramienta más usada en edición genómica deje de cortar. ¿Y si esa «debilidad» pudiera convertirse en una ventaja terapéutica?
📄 Paper: Molecular basis for methylation-sensitive editing by Cas9 · Nature, 2026
![]()
🎬 Video: [Pendiente]
Lo que midieron#
ThermoCas9 es una variante de Cas9 (de la bacteria Geobacillus thermodenitrificans) que necesita una secuencia corta de ADN llamada PAM para cortar — algo como una «matrícula de aparcamiento» que la enzima reconoce antes de actuar. Y aquí lo raro: si una citosina específica del PAM está metilada (con un grupo metilo en su carbono 5, una marca epigenética común en mamíferos), la enzima pierde casi toda su actividad.
Para probarlo atacaron por tres frentes: ensayos in vitro con ADN sintético, edición en células humanas con distintos paisajes de metilación, y cuatro fotografías a escala atómica por crio-microscopía electrónica del bolsillo que reconoce el PAM.
# ══════════════════════════════════════════════════════════════
# Configuración — modifica estos valores para explorar
# ══════════════════════════════════════════════════════════════
KI_RATIO_OBSERVADO = 12 # cuántas veces más fuerte se une al ADN no metilado
INDEL_UMBRAL = 5 # % de edición que consideramos "actividad real"
COLOR_NOMET = "#2563EB" # azul CaM — ADN sin metilar
COLOR_MET = "#DC2626" # rojo — ADN metilado
COLOR_CANCER = "#7C3AED" # violeta — línea celular cancerosa
COLOR_NORMAL = "#059669" # verde — línea celular normal
FUENTE = "Fuente: Pacesa et al. (2026), Nature | Datos: Supplementary del paper"
# ══════════════════════════════════════════════════════════════
# Setup
# ══════════════════════════════════════════════════════════════
import os, urllib.request
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
# Estilo CaM (local → fallback GitHub raw)
style_file = "../../cam.mplstyle"
if not os.path.exists(style_file):
style_file = "/tmp/cam.mplstyle"
if not os.path.exists(style_file):
urllib.request.urlretrieve(
"https://raw.githubusercontent.com/Ciencia-a-Mordiscos/lab/main/cam.mplstyle",
style_file,
)
plt.style.use(style_file)
os.makedirs("figuras", exist_ok=True)
# ══════════════════════════════════════════════════════════════
# Carga de datos
# ══════════════════════════════════════════════════════════════
indel = pd.read_csv("datos/indel_cellular.csv")
ki = pd.read_csv("datos/ki_oligos.csv")
construct = pd.read_csv("datos/construct_editing.csv")
cryoem = pd.read_csv("datos/cryoem_resolutions.csv")
print(f"indel: {len(indel)} mediciones · {indel['site'].nunique()} sitios × {indel['cell_line'].nunique()} líneas")
print(f"ki: {len(ki)} condiciones (ratio metilado/no metilado = {ki['ki_nm'].iloc[1]/ki['ki_nm'].iloc[0]:.1f}×)")
print(f"construct: {len(construct)} condiciones (MCF-7 cáncer vs MCF-10A normal)")
print(f"cryoem: {len(cryoem)} estructuras · resolución {cryoem['resolution_angstrom'].min()}–{cryoem['resolution_angstrom'].max()} Å")
indel: 8 mediciones · 4 sitios × 2 líneas
ki: 2 condiciones (ratio metilado/no metilado = 12.0×)
construct: 6 condiciones (MCF-7 cáncer vs MCF-10A normal)
cryoem: 4 estructuras · resolución 2.2–3.5 Å
Aquí está.#
# Indel por estado de metilación del PAM (4 sitios × 2 líneas celulares)
fig, ax = plt.subplots(figsize=(13, 5.5))
np.random.seed(42)
nomet = indel[indel["pam_methylation_status"] == "unmethylated"]["indel_pct"].values
met = indel[indel["pam_methylation_status"] == "methylated"]["indel_pct"].values
x_nomet = np.linspace(-0.18, 0.18, len(nomet))
np.random.shuffle(x_nomet)
x_met = np.linspace(-0.18, 0.18, len(met))
np.random.shuffle(x_met)
ax.scatter(x_nomet, nomet, color=COLOR_NOMET, s=120, alpha=0.85,
edgecolors="white", linewidths=1.2, zorder=5)
ax.scatter(1 + x_met, met, color=COLOR_MET, s=120, alpha=0.85,
edgecolors="white", linewidths=1.2, zorder=5)
mean_nomet, sem_nomet = nomet.mean(), nomet.std(ddof=1) / np.sqrt(len(nomet))
mean_met = met.mean()
sem_met = met.std(ddof=1) / np.sqrt(len(met)) if met.std() > 0 else 0
ax.errorbar(0, mean_nomet, yerr=sem_nomet, fmt="_", color=COLOR_NOMET,
markersize=22, markeredgewidth=3, capsize=8, capthick=2, zorder=6)
ax.errorbar(1, mean_met, yerr=sem_met, fmt="_", color=COLOR_MET,
markersize=22, markeredgewidth=3, capsize=8, capthick=2, zorder=6)
ax.set_xticks([0, 1])
ax.set_xticklabels(["PAM sin metilar", "PAM metilado"], fontsize=11, fontweight="bold")
ax.get_xticklabels()[0].set_color(COLOR_NOMET)
ax.get_xticklabels()[1].set_color(COLOR_MET)
ax.set_ylabel("Frecuencia de indel (%)", fontsize=11)
ax.set_ylim(-3, 38)
ax.set_xlim(-0.6, 1.6)
ax.set_title("¿Qué pasa cuando el PAM está metilado?",
fontsize=14, fontweight="bold", pad=28)
ax.text(0.5, 1.03, "Edición de ThermoCas9 en HEK293T y HCT116, 4 sitios genómicos",
transform=ax.transAxes, fontsize=10, color="#666666", ha="center")
ax.text(0.98, 0.02, "━ media ± SEM", transform=ax.transAxes,
fontsize=8, color="#999999", ha="right", va="bottom", style="italic")
fig.text(0.13, -0.03, FUENTE, fontsize=7.5, color="#999999", style="italic")
plt.savefig("figuras/indel_methylation.png", dpi=200, bbox_inches="tight")
plt.show()
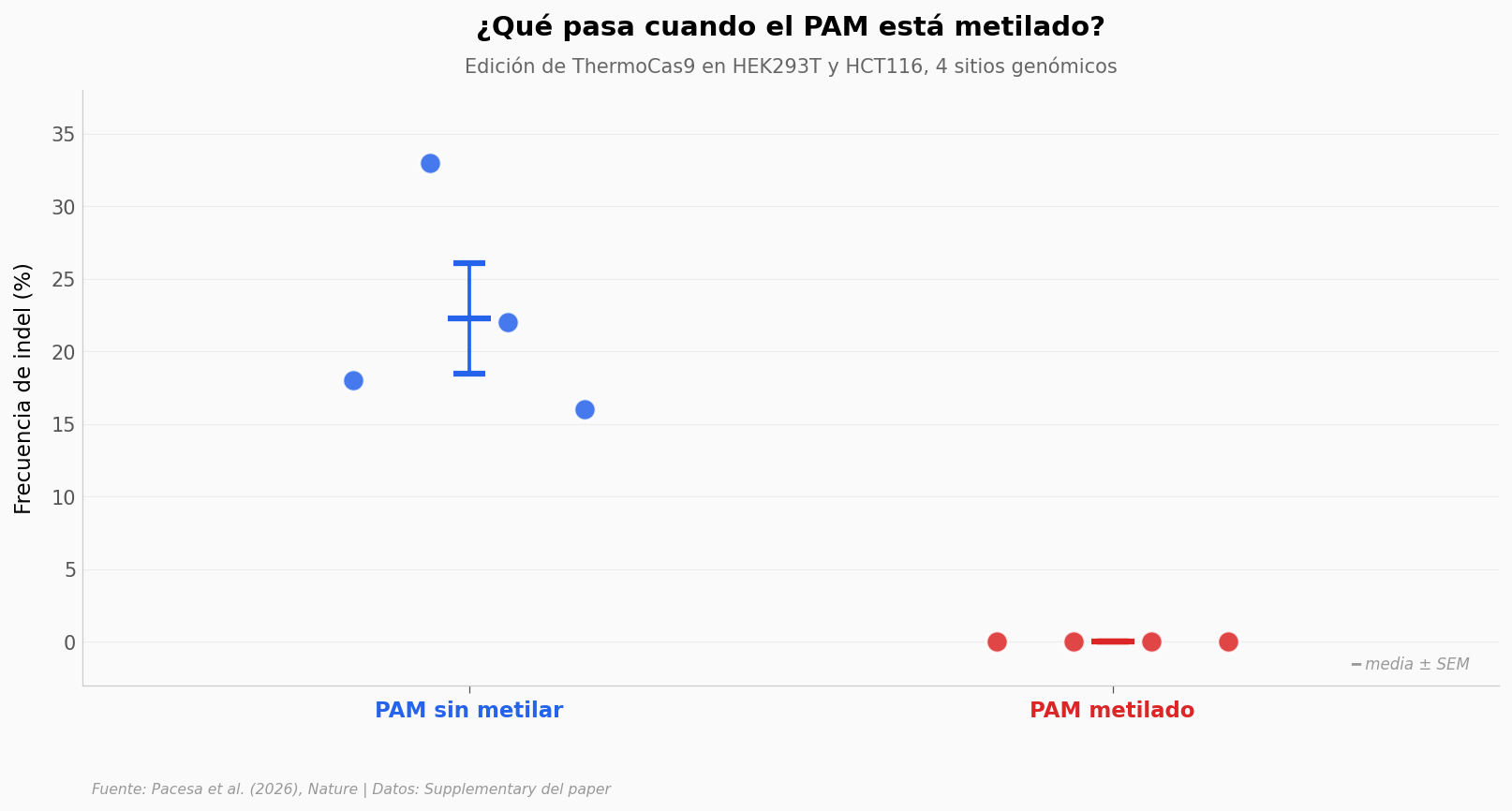
Lectura guiada#
Lo que llama la atención no es que la edición baje. Es que se apaga. Cuando el PAM está metilado, ThermoCas9 deja de cortar — la frecuencia de indel cae a cero en los cuatro sitios probados, sin importar la línea celular. Sin metilación la edición oscila entre el 16% y el 33%, valores típicos para Cas9 en cultivo.
El detalle clave: el equipo eligió pares de sitios donde la secuencia es idéntica entre líneas celulares. Lo único que cambia es el estado de metilación. Eso elimina el efecto de la cromatina, la accesibilidad y la secuencia local — la metilación es el único sospechoso.
¿Qué tan fuerte es la preferencia?#
Para cuantificar el efecto sin la complicación del entorno celular, midieron in vitro la constante de inhibición (Ki) — un número que dice cuánta concentración de un competidor se necesita para frenar a la enzima. Ki bajo = competidor fuerte; Ki alto = competidor débil.
# Ki: ADN sin metilar vs metilado (in vitro)
fig, ax = plt.subplots(figsize=(11, 5))
labels = ["ADN sin metilar", "ADN metilado"]
values = ki["ki_nm"].values
errors = ki["ki_se_nm"].values
colors = [COLOR_NOMET, COLOR_MET]
bars = ax.barh(labels, values, xerr=errors, color=colors, alpha=0.85,
edgecolor="white", linewidth=1.5,
error_kw={"capsize": 6, "capthick": 1.5, "elinewidth": 1.5})
for i, (v, e) in enumerate(zip(values, errors)):
ax.text(v + e + 25, i, f"Ki = {v} ± {e} nM",
fontsize=11, fontweight="bold", color=colors[i], va="center")
ax.set_xlabel("Constante de inhibición Ki (nM) — más bajo = mejor competidor", fontsize=10)
ax.set_xlim(0, 1100)
for tick, color in zip(ax.get_yticklabels(), colors):
tick.set_color(color)
tick.set_fontweight("bold")
ax.set_title("ThermoCas9 prefiere ADN sin metilar 12 veces más",
fontsize=14, fontweight="bold", pad=28)
ax.text(0.5, 1.03, "Ki medido por ensayos de competición con oligonucleótidos sintéticos",
transform=ax.transAxes, fontsize=10, color="#666666", ha="center")
ax.annotate("", xy=(767, 0.7), xytext=(64, 0.7),
arrowprops=dict(arrowstyle="<->", color="#666666", lw=1.5))
ax.text((64 + 767) / 2, 0.55, "12×", fontsize=14, fontweight="bold",
color="#666666", ha="center")
fig.text(0.13, -0.05, FUENTE, fontsize=7.5, color="#999999", style="italic")
plt.savefig("figuras/ki_oligos.png", dpi=200, bbox_inches="tight")
plt.show()
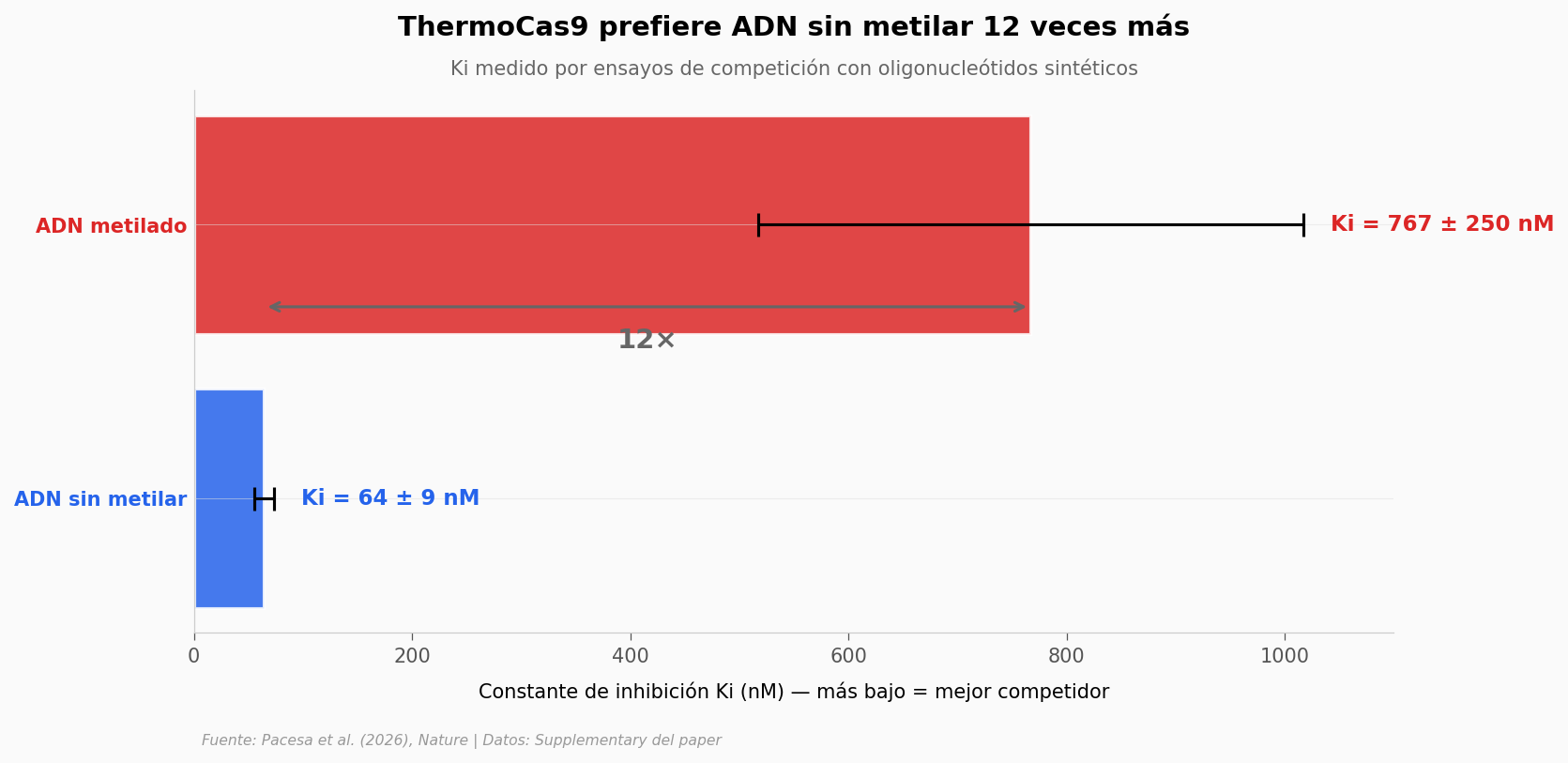
La grieta terapéutica#
Si la enzima distingue ADN metilado de ADN no metilado, entonces edita preferentemente células que tienen sus genes diana hipo-metilados. Y eso es justo lo que pasa con ciertos genes en cáncer de mama: en tumores luminales, GATA3 y ESR1 — genes que controlan la identidad celular — pierden su metilación normal mientras siguen metilados en células sanas.
El equipo lo probó en una pareja clásica del laboratorio: MCF-7 (línea de cáncer de mama, hipometilada en estos sitios) y MCF-10A (línea epitelial mamaria normal, metilada).
# Edición en MCF-7 (cáncer) vs MCF-10A (normal) con ThermoCas9 catalíticamente reforzado (CE-RNP)
fig, ax = plt.subplots(figsize=(12, 5.5))
df = construct[(construct["construct"] == "CE_RNP") & construct["pct"].notna()].copy()
df["label"] = df["cell_line"] + " · " + df["site"]
df = df.sort_values("pct", ascending=True).reset_index(drop=True)
df["color"] = df["cell_line"].map({"MCF-7": COLOR_CANCER, "MCF-10A": COLOR_NORMAL})
ax.barh(df["label"], df["pct"], color=df["color"], alpha=0.85,
edgecolor="white", linewidth=1.5)
for i, (lab, p) in enumerate(zip(df["label"], df["pct"])):
ax.text(p + 1.5, i, f"{p:.0f}%", fontsize=10, fontweight="bold",
color=df["color"].iloc[i], va="center")
ax.set_xlabel("Reads modificadas (%)", fontsize=10)
ax.set_xlim(0, 90)
ax.text(82, len(df) - 0.5, "MCF-7 (cáncer, hipometilado)", fontsize=10,
color=COLOR_CANCER, fontweight="bold", ha="right", va="center")
ax.text(82, 0.5, "MCF-10A (normal, metilado)", fontsize=10,
color=COLOR_NORMAL, fontweight="bold", ha="right", va="center")
ax.set_title("¿Qué pasa cuando la línea es de cáncer vs normal?",
fontsize=14, fontweight="bold", pad=28)
ax.text(0.5, 1.03, "ThermoCas9 catalíticamente reforzado (CE-RNP) sobre genes luminales",
transform=ax.transAxes, fontsize=10, color="#666666", ha="center")
fig.text(0.13, -0.05, FUENTE, fontsize=7.5, color="#999999", style="italic")
plt.savefig("figuras/mcf7_vs_mcf10a.png", dpi=200, bbox_inches="tight")
plt.show()
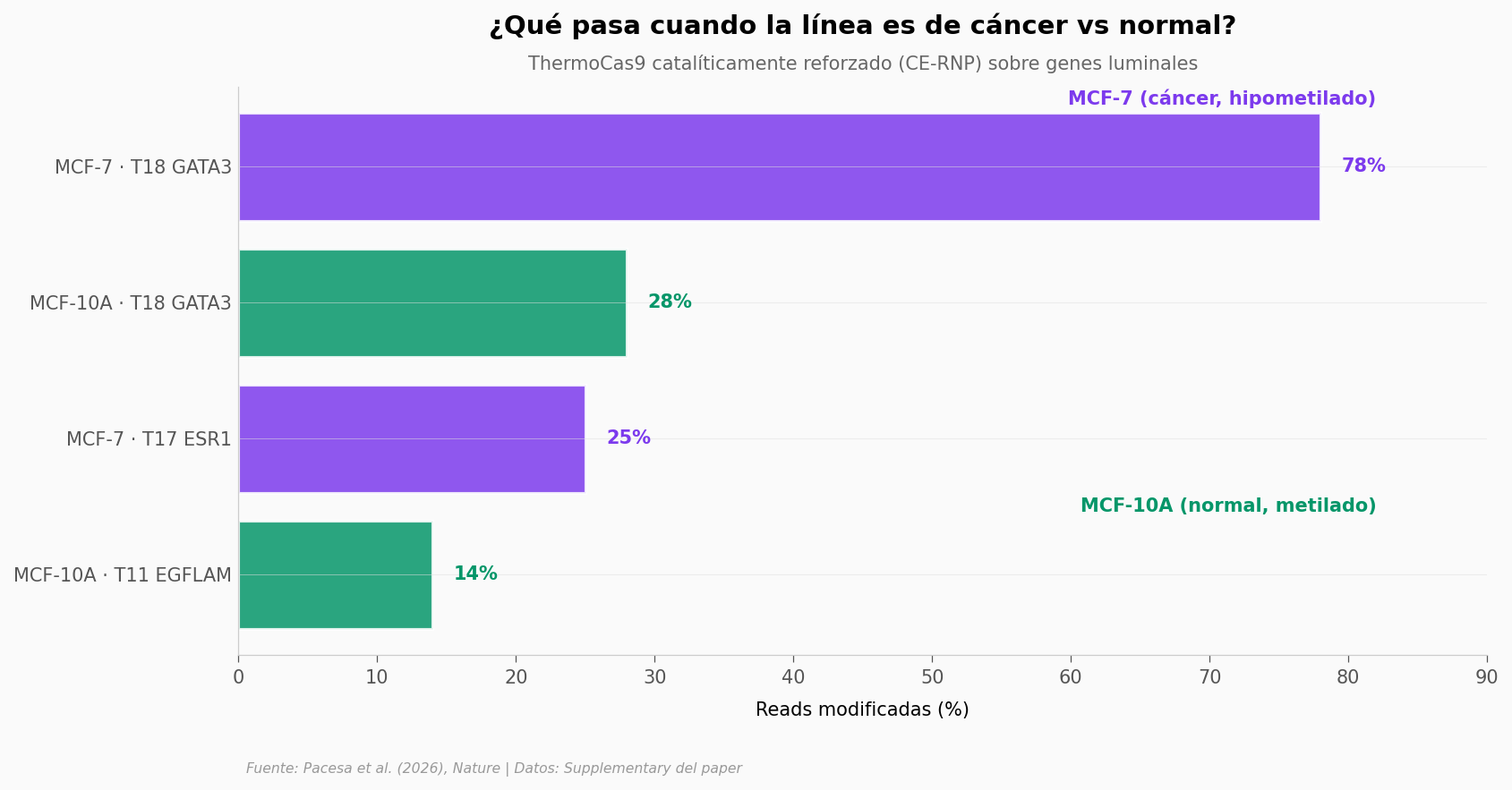
¿Cómo lo ven a escala atómica?#
Para entender el mecanismo a fondo había que ir a escala atómica. Cuatro estructuras de crio-microscopía electrónica, la mejor a 2,2 Å — nítida como para ver el grupo metilo individual y el aminoácido que lo rechaza.
# Resolución de las 4 estructuras de crio-EM
fig, ax = plt.subplots(figsize=(11, 5))
df = cryoem.sort_values("resolution_angstrom").copy().reset_index(drop=True)
ax.barh(df["pdb_id"], df["resolution_angstrom"],
color=COLOR_NOMET, alpha=0.85, edgecolor="white", linewidth=1.5)
for i, (pdb, res, desc) in enumerate(zip(df["pdb_id"], df["resolution_angstrom"], df["description"])):
ax.text(res + 0.05, i, f"{res} Å · {desc}",
fontsize=9, color="#444444", va="center")
ax.set_xlabel("Resolución (Å) — más bajo = más detalle atómico", fontsize=10)
ax.set_ylabel("Código PDB", fontsize=10)
ax.set_xlim(0, 5.5)
ax.axvline(x=3.0, color="#999999", linewidth=1, linestyle="--", alpha=0.7)
ax.text(3.05, -0.6, "≈ 3 Å: detalle de cadena lateral",
fontsize=8, color="#999999", style="italic")
ax.set_title("Cuatro fotografías a escala atómica",
fontsize=14, fontweight="bold", pad=28)
ax.text(0.5, 1.03, "Estructuras depositadas en PDB y EMDB con sus resoluciones",
transform=ax.transAxes, fontsize=10, color="#666666", ha="center")
fig.text(0.13, -0.05, FUENTE, fontsize=7.5, color="#999999", style="italic")
plt.savefig("figuras/cryoem_resolutions.png", dpi=200, bbox_inches="tight")
plt.show()
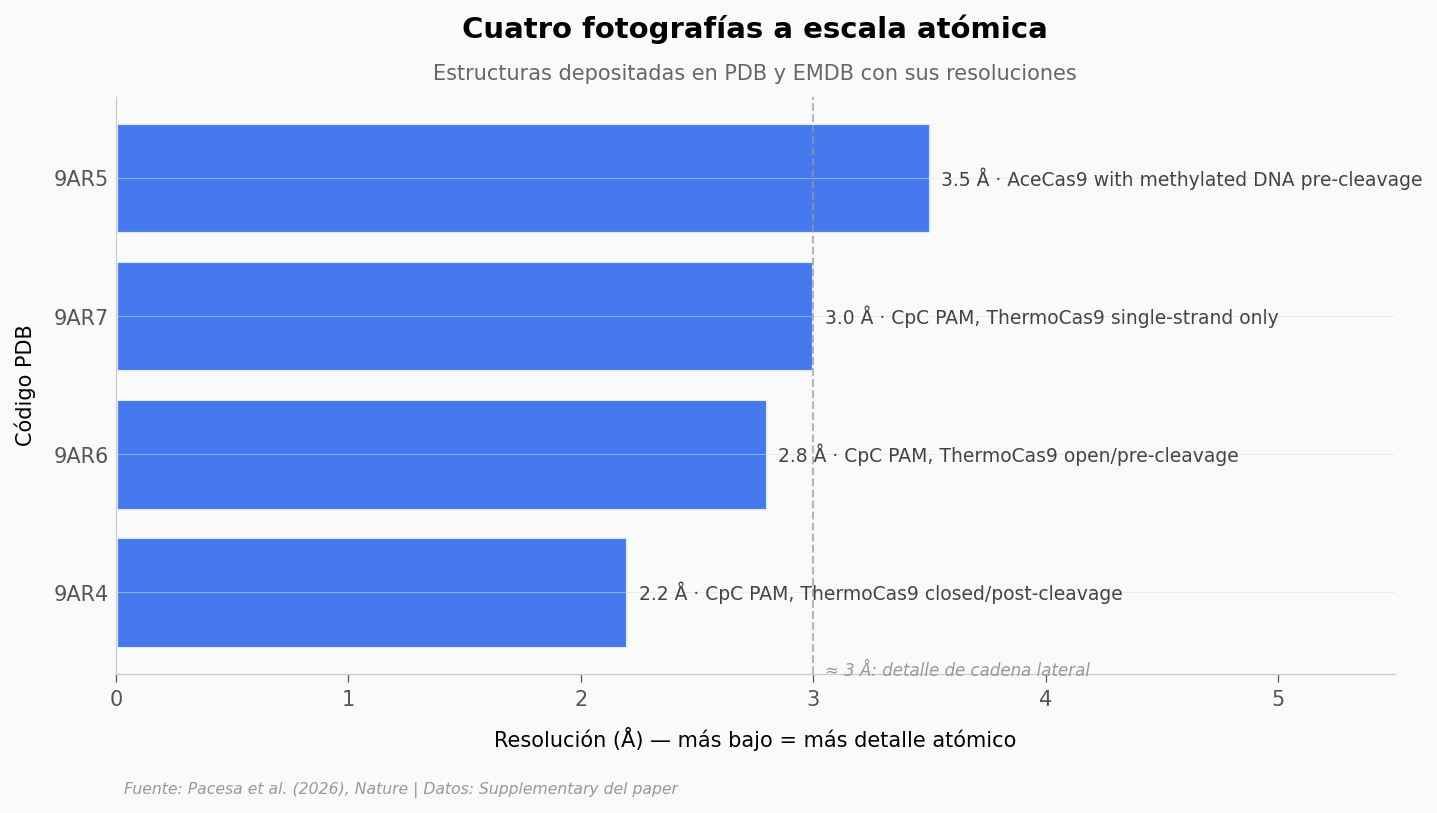
Lo que los datos soportan#
Afirmación |
¿Soportada? |
Detalle |
|---|---|---|
La metilación del PAM apaga la edición |
✅ |
4/4 sitios metilados → 0% indel; 4/4 sitios sin metilar → 16-33%. Diseño cruzado por línea celular controla efectos de cromatina. |
ThermoCas9 prefiere 12× ADN sin metilar in vitro |
✅ |
Ki = 64 ± 9 nM (sin metilar) vs 767 ± 250 nM (metilado). El error en el metilado es alto (32% relativo) — el ratio es robusto pero el valor absoluto tiene incertidumbre. |
MCF-7 (cáncer) edita más que MCF-10A (normal) |
⚠️ |
MCF-7 GATA3 alcanza 78%, MCF-10A 14-28%. Sin embargo, el control en MCF-10A no es 0% — la ventana terapéutica existe pero no es absoluta. |
4 estructuras de crio-EM con resoluciones 2,2-3,5 Å |
✅ |
Depositadas en PDB (9AR4-9AR7) y EMDB (43769-43772). La de 2,2 Å permite ver el grupo metilo. |
Aplicación clínica directa |
⚠️ |
El paper enmarca esto como muestra potencial para terapias. No hay datos in vivo ni en pacientes. |
Limitaciones del análisis: N pequeño (4 sitios, 2 líneas celulares); no recalculamos test estadístico — el paper reporta two-way ANOVA con corrección Sidak (p<0.0001). Los Ki tienen errores estándar grandes (especialmente en metilado: ±250 sobre 767, 32% relativo). Las líneas celulares MCF-7 y MCF-10A difieren en más cosas que la metilación (genoma, contexto).
Ahora tú#
Tres preguntas para cacharrear con los datos:
¿Cuál sería el ratio de Ki si los errores estándar fueran sumados (peor caso)? Pista: usa el percentil más alto del intervalo metilado y el más bajo del no metilado.
¿Hay alguna ventaja consistente entre MCF-7 y MCF-10A para los sitios donde ambas tienen datos? Pista: filtra
construct[construct["construct"] == "CE_RNP"]y compara por sitio.¿La diferencia entre HEK293T y HCT116 es mayor que la diferencia metilado/no metilado? Pista: agrupa
indelporcell_liney compara las medias.
# --- EXPERIMENTA AQUÍ ---
# Pregunta 1: ratio en el peor caso (Ki metilado más bajo / Ki no metilado más alto)
unmet = ki[ki["oligo_type"] == "unmethylated_oligo"].iloc[0]
methy = ki[ki["oligo_type"] == "target_strand_methylated_oligo"].iloc[0]
ki_unmet_alto = unmet["ki_nm"] + unmet["ki_se_nm"]
ki_met_bajo = methy["ki_nm"] - methy["ki_se_nm"]
print(f"Ki sin metilar (cota alta): {ki_unmet_alto} nM")
print(f"Ki metilado (cota baja): {ki_met_bajo} nM")
print(f"Ratio en el peor caso: {ki_met_bajo/ki_unmet_alto:.1f}×")
print(f"Ratio reportado (medias): 12.0×")
print()
print("Incluso en el peor escenario, la preferencia se mantiene clara —")
print("aunque pasa de 12× a un múltiplo más modesto.")
Ki sin metilar (cota alta): 73 nM
Ki metilado (cota baja): 517 nM
Ratio en el peor caso: 7.1×
Ratio reportado (medias): 12.0×
Incluso en el peor escenario, la preferencia se mantiene clara —
aunque pasa de 12× a un múltiplo más modesto.
Fuentes#
Paper: Molecular basis for methylation-sensitive editing by Cas9
Nature, 2026-04-15
Datos: Supplementary Information, Extended Data Tables y estructuras depositadas en PDB (9AR4–9AR7) y EMDB (EMD-43769 a EMD-43772)
14 afirmaciones verificadas contra estas fuentes
📂 Repo: github.com/Ciencia-a-Mordiscos/lab · Licencia: CC BY 4.0
🔬 Notebook reproducible: jupyter execute notebook.ipynb